Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Кома с лактат-ацидозом (молочнокислая)
|
|
Это относительно редкое, но опасное осложнение сахарного диабета. В механизме ее развития важную роль играют следующие факторы:
а) снижение активности фермента пируват-
дегидрогеназы (наблюдается при дефиците ин
сулина), превращающего пируват в ацетил-КоА.
Пируват в обратимой реакции, катализируемой
лактатдегдрогеназой, превращается в молочную
кислоту;
б) применение лекарственных препаратов,
стимулирующих анаэробный гликолиз и, тем
самым, повышающих содержание лактата и пи-
рувата в организме (например, бигуаниды, по
вышающие утилизацию глюкозы за счет ее ана
эробного распада). При поражении печени или
почек может наблюдаться кумуляция этих пре
паратов в организме, в результате чего развива
ется лактат-ацидоз и кома;
в) гипоксическое состояние (при котором, как
правило, стимулируется гликолиз), вызванное
физическим переутомлением, сердечной или
дыхательной недостаточностью.
Как следствие, в крови накапливается молочная кислота (содержание лактата в плазме превышает 5 ммоль/л), что сопровождается развитием коллапса, нарушением сердечной деятельности и функций дыхательного центра (возникает патологическое дыхание Куссмауля), угнетением сознания, нарушением чувствительности, дисфункцией желудочно-кишечного тракта, резко выраженной дегидратацией тканей. Гипер-кетонемия и кетонурия отсутствуют, могут наблюдаться незначительная гипергликемия и небольшая глюкозурия. Вследствие несвоевременной диагностики и трудности лечения прогноз может быть неблагоприятным.
4.Гипогликемическая кома. Возникающая при сахарном диабете, она связана с передозировкой инсулина, развитием вторичного гипо-питуитаризма (следствие ангиопатии сосудов гипофиза), ослабляющего ответ на гипогликемию, и явлениями диабетического нефроскле-роза, что удлиняет время циркуляции инсулина и, кроме того, еще более снижает почечный порог для глюкозы, способствуя ее потере.
Причинами гипогликемии могут быть также гиперпродукция инсулина опухолью поджелудочной железы (инсулиномой), недостаточность контринсулярных гормонов, гликогенозы, заболевания печени, голодание, нарушение расщеп-Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
ления и всасывания углеводов в желудочно-кишечном тракте и др.
В механизме развития гипогликемической комы решающее значение имеет снижение доставки глюкозы к нервным клеткам, что ведет к их энергетическому истощению и нарушению функций ЦНС. При снижении уровня глюкозы менее 3 ммоль/л возникают потливость, тремор, чувство тревоги и голода, слабость. Затем развивается состояние, напоминающее алкогольное опьянение и сопровождающееся дезориентацией, агрессивностью, галлюцинациями. При дальнейшем падении содержания глюкозы (менее 2,5 ммоль/л) возникают клонические судороги и потеря сознания. В тяжелых случаях могут наступать отек и некроз отдельных участков мозга.
Поздние осложнения диабета
Поздние осложнения диабета являются основными причинами большей части смертельных исходов заболевания. К ним относятся:
1. Макроангиопатия. У больных сахарным диабетом атеросклероз развивается существенно раньше, чем у здоровых людей. Патологический процесс охватывает сосуды головного мозга, сердца, почек, а также сосуды конечностей, в особенности сосуды голени и стопы. Диабет, даже в условиях его лечения современными средствами, характеризуется ускоренными темпами старения организма. Наличием диабета обусловлена высокая частота инфарктов миокарда, инсультов и случаев гангрены пальцев ног или стопы. В настоящее время считают, что диабет ускоряет развитие атеросклероза в результате:
а) избытка гормона роста (и отсутствия про
тиводействия со стороны инсулина в условиях
его абсолютного или относительного дефицита),
приводящего к усилению процесса пролифера
ции гладкомышечных клеток;
б) усиленного синтеза тромбоксана, способ
ствующего увеличению адгезии тромбоцитов и
выделению ряда факторов, которые также усу
губляют темпы пролиферации и миграции глад
комышечных клеток;
в) стойкого увеличения концентрации в кро
ви липопротеидов низкой плотности (ЛПНП) при
одновременном снижении таковой для липопро
теидов высокой плотности (ЛПВП).
Несомненный интерес представляют данные о наследственной предрасположенности больных
диабетом к атеросклерозу. Установлено, что рядом с геном, кодирующим синтез инсулина, расположен участок ДНК (U-аллель) - постоянный генетический маркер предрасположенности к атеросклерозу не только у больных диабетом I и II типов, но также и у лиц без диабета. Однако у больных диабетом наследственная предрасположенность к атеросклерозу реализуется чаще, чем у лиц без диабета.
2. Микроангиопатия. Это осложнение выражается в повреждении капиллярной сети (и примыкающих к ней сосудов), чаще всего поражая почки и сетчатку глаз.
Поражение почек (диабетическая нефропа-тия) вследствие развития макро- и микроангио-патий в настоящее время является основной причиной ранней смертности у диабетиков молодого возраста. При этом происходит избыточное гликозилирование коллагена базальных мембран почечных клубочков, приводящее к существенным нарушениям структуры и функций этих мембран (рис. 87).
Если в суточной моче концентрация альбумина выше 30 мг и эти значения повторяются несколько раз, то необходимо проводить лечение, так как данные изменения характерны для начинающейся диабетической нефропатии.
По мере прогрессирования поражения почек при диабете развивается выраженная протеину-рия. Тщательный контроль за уровнем глюкозы в крови и лечение любых форм гипертонии может приостановить микроальбуминемию и предупредить развитие манифестной почечной недостаточности.
Сиалогликопротеины
Просвет канальца
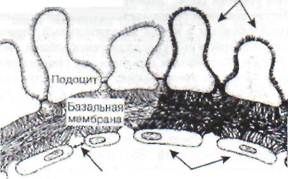
Фенестры Эндотелиальные клетки
Просвет капилляра Рис. 87. Гликозилирование поверхностных белков мембран эндотелиальных и эпителиальных клеток
(подоцитов) с потерей поверхностного заряда при диабете
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
I') ЗавпИЬ 5.12
Проявления избыточного гликозилирования белков при сахарном диабете
Таблица 40
| Белки | Патологические проявления |
| Белки мембран эритроцитов Белки свертывающей системы крови Белки клеточных мембран эндотелия Белки хрусталика и его капсулы Белки базальных мембран клеток почечных клубочков Коллаген Миелин Переносчик глюкозы | Деформация эритроцитов Нарушения свертываемости крови Нарушения проницаемости сосудов Нарушения зрения Патология почечных клубочков Нарушения рубцевания ран Патология нервной системы Инсулиновая резистентность |
Поражение сетчатки глаз при диабете (диабетическая ретинопатия) относится к числу одной из наиболее частых причин развивающейся слепоты при этой патологии. Длительно существующая гипергликемия вызывает усиление синтеза сорбитола и фруктозы. Накопление этих углеводов в хрусталике глаза вызывает (по осмотическому механизму) увеличение содержания в нем воды, что обусловливает необратимые нарушения структуры хрусталика - формируется диабетическая катаракта.
3. Гликозилированный гемоглобин и другие белки. Гликозилированный гемоглобин (НЬА1с) - минорный компонент гемоглобина, отличающийся от гемоглобина основного вида - НЬА. Избыточное количество НВА1с формируется благодаря длительной гипергликемии: неферментативным путем образуются ковалентные связи между молекулами глюкозы и N-концевыми остатками аминокислоты валина Р-цепей НЬА.
Установлено, что скорость образования НЬА1с пропорциональна произведению концентрации глюкозы в эритроцитах на время. На этом основании уровень НЪА]с может быть использован в качестве косвенного показателя средней концентрации глюкозы в крови за длительный период времени (период полураспада гемоглобина около 60 сут). Косвенным показателем гипергликемии за меньшее время может служить гликозилированный альбумин (период полураспада альбумина около 20 сут).
Избыточное гликозилирование других белков может играть определенную роль в патогенезе некоторых нарушений, обусловленных поздними осложнениями диабета. Неферментативному гликозилированию при длительной гипергликемии могут подвергаться как структурные белки, так и ферменты. Примеры таких белков приведены в табл. 40.
Особый интерес представляют последствия
гликозилирования белков (апопротеинов) сывороточных липопротеидов. Так, процесс, затрагивающий апопротеины липопротеидов низкой плотности, приводит к нарушению взаимодействия ЛПНП с их рецепторами на плазматических мембранах клеток и, как следствие, замедляется удаление ЛПНП из кровотока. В результате увеличивается концентрация холестерина в крови.
Гликозилирование апопротеинов, входящих в состав липопротеидов высокой плотности, которые транспортируют холестерин из периферических тканей в печень, приводит к ускорению удаления ЛПВП из кровотока. При этом в крови увеличивается соотношение ЛПНП/ЛПВП.
4. Диабетическая нейропатия. Нейропатии (нарушения функции нервов) способны вызывать дисфункции любой системы организма, имитируя многочисленные неврологические заболевания. В патологический процесс могут быть вовлечены как чувствительные, так и двигательные или вегетативные нервные волокна. В качестве типичных примеров клинического проявления диабетических нейропатии можно назвать образование язв на стопах, различные расстройства функций желудочно-кишечного тракта, мочевого пузыря, импотенцию и др. При исследовании структуры нервных волокон диабетиков с помощью световой микроскопии часто выявляются признаки их демиелинизации, хотя нарушения функции волокон на фоне этих явлений клинически могут и не проявляться.
Патогенез диабетических нейропатии полностью не раскрыт, однако в настоящее время можно назвать ряд факторов, безусловно определяющих развитие этого осложнения диабета:
а) нарушение структуры миелина. Патологический процесс обусловливает изменение как химической структуры, так и количественного соотношения между основными биохимически-
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
ми компонентами (холестерин, триацилглицери-ды, фосфолипиды, гликолипиды и белки) миелина нервных волокон. Клинические наблюдения свидетельствуют о том, что под влиянием заместительной терапии инсулином происходит существенная коррекция многих из указанных сдвигов;
б) сорбитоловый путь окисления глюкозы. В результате данного процесса происходит ферментативное окисление глюкозы с образованием сначала сорбитола, а затем фруктозы, повышенное количество которых формирует осмотические сдвиги во внутриклеточном пространстве тканей. Одним из следствий данного феномена является снижение потребления кислорода нервной тканью, что, вероятно, лежит в основе нарушений ее функционирования. В условиях снижения гипергликемии упомянутые дисфункции обратимы.
Следует отметить, что общая черта всех вышеперечисленных нарушений, по крайней мере до развития макроморфологических изменений, таких как демиелинизация, - их обратимость под влиянием терапии, приводящей к снижению гипергликемии. С помощью определенных методик введения инсулина больным диабетом, максимально имитирующих характер и режим секреции гормона (3-клетками островкового аппарата поджелудочной железы, возможно если не предупредить многие осложнения диабета, то хотя бы существенно задержать их развитие.
Обнаружение нарушений углеводного обмена методом нагрузок (тесты толерантности к глюкозе)
Исследование состояния углеводного обмена с диагностической целью в клинике начинают с определения натощак содержания сахара в крови и анализа мочи на присутствие в ней сахара и кетоновых тел. Если результаты анализов свидетельствуют о наличии гипергликемии, глюкозо- и кетонурии, то этого оказывается достаточно для подтверждения диагноза сахарного диабета. Доказано, что ни одно из других заболеваний внутренних органов не дает всей триады: гипергликемии, глюкозо- и кетонурии. Лишь при сахарном диабете имеют место существенные нарушения не только углеводного, но и жирового обмена.
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕН
В случае, если результаты упомянутых анализов крови и мочи дают норму или незначительное ее превышение, то более углубленное исследование состояния углеводного обмена производят при помощи сахарной нагрузки.
Проба с однократной сахарной нагрузкой для получения гликемической («сахарной») кривой. У обследуемого утром натощак берут кровь из пальца для определения концентрации глюкозы, после чего дают сахарную нагрузку: прием внутрь 100 г глюкозы, растворенной в 200 мл кипяченой воды. Время, в течение которого раствор следует выпить, не должно превышать 5 мин. Повторные заборы проб крови из пальца ведут с интервалом в 30 или 60 мин. Длительность пробы (у взрослых) составляет 3 ч. На основании полученных данных строят кривую, откладывая по оси ординат концентрацию глюкозы, а по оси абсцисс - время. Типы гликеми-ческих кривых, присущих норме или сахарному диабету, представлены на рис. 88.
Для гликемической кривой у здоровых субъектов характерны следующие признаки. Уже через 15 мин после приема раствора глюкозы внутрь в крови начинает расти концентрация глюкозы, достигая максимума к концу первого часа (в промежутке от 30-й до 60-й мин). При этом концентрация превышает таковую натощак на 50-75%. Далее концентрация глюкозы в крови начинает снижаться и к концу второго часа наблюдения (к 120-й мин) она либо достигает исходного уровня (натощак), либо падает ниже исходного уровня (вариант физиологической гипогликемии, см. разд. 11.4.5), либо остается несколько повышенной, но не превышает значения 6,6 ммоль/л. К третьему часу во всех трех возможных вариантах концентрация глюкозы в крови не отличается от исходного значения (натощак).
У больных сахарным диабетом концентрация глюкозы натощак повышена, а нарастание гликемической кривой после сахарной нагрузки происходит медленнее. Максимальное значение показателя регистрируют только через 60-150 мин от начала наблюдения, при этом концентрация глюкозы может в 1,8 раза превышать ее исходное значение. Спад концентрации глюкозы крови (гипогликемическая фаза) также происходит чрезвычайно медленно (вплоть до отсутствия такового), что коррелирует со степенью тяжести заболевания. Если же понижение
Й ОБМЕНА ВЕЩЕСТВ 291
концентрации глюкозы все же происходит, то оно растягивается на 3-4 ч.
Для трактовки гликемических кривых (на основе оценки высоты подъема концентрации глюкозы в крови после нагрузки и характера ее падения) вычисляются различные коэффициенты. Например:
а) гликемический коэффициент Водуэна:
Кл = В/А,
Ьодуэня ' *
где В - концентрация глюкозы в максимуме подъема; А - исходный уровень показателя (натощак).
В норме величина данного коэффициента составляет 1,3 - 1,5;
б) постгликемический коэффициент Рафаль-
ского
^Рафальского = WAr
где С • самая низкая концентрация глюкозы в крови (определяемая через 2 ч после нагрузки); А - исходный уровень показателя (натощак).
В норме величина этого коэффициента составляет 0,9 - 1,04.
Считается, что решающую диагностическую информацию дает не столько величина приведенных коэффициентов, сколько тип гликеми-ческой кривой.
Тест толерантности к глюкозе. Кровь для проведения теста также берут из пальца дважды: натощак и спустя 120 мин после нагрузки глюкозой. Согласно критериям ВОЗ у практически здорового человека концентрация глюкозы в крови натощак не должна превышать 5,55
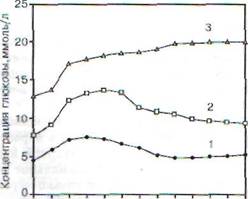
60 90 120 150 180
Время,мин
Рис. 88. Типы гликемических кривых в норме и в
состояниях, характеризующихся пониженной
толерантностью к глюкозе: / - норма; 2 - латентный
сахарный диабет (легкая форма заболевания);
3 - тяжелая форма сахарного диабета
ммоль/л. Спустя 120 мин после стандартной нагрузки глюкозой (одномоментный прием внутрь раствора глюкозы из расчета 1 г глюкозы на 1 кг массы) уровень глюкозы не должен превышать 6,66 ммоль/л. В случае, если концентрация глюкозы натощак превышает 7,22 ммоль/л, а тест свидетельствует о том, что спустя 120 мин уровень глюкозы остается выше 7,77 ммоль/л, то это веский аргумент в пользу если не диабета, то преддиабетического состояния у обследуемого.
11.4.5. Гипогликемические состояния
Гипогликемия - снижение уровня глюкозы в крови ниже 3,3 ммоль/л. Причинами гипогликемии могут быть недостаточное поступление глюкозы в кровь, ускоренное выведение ее из крови либо комбинация этих факторов.
Физиологическая гипогликемия. Наблюдается при тяжелой и длительной физической нагрузке; у женщин в период лактации; развивается сразу вслед за алиментарной гипергликемией благодаря компенсаторному выбросу в кровь инсулина.
Патологическая гипогликемия (гиперинсу-линизм). Чаще возникает у больных сахарным диабетом в связи с передозировкой инсулина при лечении. Причиной ее могут быть также: аденома островковых клеток поджелудочной железы; синдром Золлингера - Эллисона (аденома или карцинома поджелудочной железы, которая, по-видимому, развивается из сс-клеток островков Лангерганса, ответственных за выделение глю-кагона и гастрина).
Патологическая гипогликемия (без гиперин-сулинизма). Встречается при: патологии почек, сопровождающейся снижением порога для глюкозы, что приводит к потере глюкозы с мочой; нарушении всасывания углеводов; заболеваниях печени, сопровождающихся торможением синтеза гликогена и глюконеогенеза (острые и хронические гепатиты); недостаточности надпочечников (дефицит глюкокортикоидов); галак-тоземиях и при некоторых типах гликогенозов; голодании или недостаточном питании (алиментарная гипогликемия); несовершенности механизмов регуляции углеводного обмена у новорожденных.
Центральная нервная система особенно чувствительна к дефициту глюкозы, поскольку именно глюкоза служит для этой ткани основным источником энергии. Малая доля потреб-
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
Таблица 41 Наиболее важные и часто встречающиеся жирные кислоты (по М.А. Карабасовой, 1997)
Примечание. Код обозначает число углеродных атомов (С20) и число двойных связей (:5), т.е. С20:5, нередко код кислоты включает и положение терминальной (омега) связи - С20:5(о-3 (так условно записывается эикозопентаеновая кислота).
| к незаменимым и условно объединены в группу под названием витамин F. Холестеринотносится к стероидным спиртам. Он является источником образования желчных кислот, стероидных гормонов, витамина D, входит в состав клеточных мембран, является важным компонентом липопротеидов плазмы крови. Фосфолипиды- это сложные эфиры многоатомных спиртов с высшими жирными кислотами и фосфорной кислотой, в их состав входят азотсодержащие соединения: холин, этаноламин, серии. Они содержатся в мембранах клеток и клеточных органелл, регулируют их проницаемость и активность Na'/K' - АТФазы, К' -АТФазы, Са24 - АТФазы, аденилатциклазы и др. Патология обмена липидов связана с нарушением их расщепления, всасывания, транспорта, утилизации, депонирования и метаболизма. 11.5.1. Нарушение переваривания и всасывания липидов Для нормального переваривания и всасывания ихв кишечнике определяющее значение имеет взаимодействие таких факторов, как: |
ности в энергии может покрываться за счет окисления кетоновых тел.
11.5. НАРУШЕНИЕ ОБМЕНА ЛИПИДОВ
Липиды - это химические соединения, нерастворимые в воде, но растворимые в хлороформе или спирте. К липидам относятся ненасыщенные и насыщенные жирные кислоты, моно-, ди-, триацилглицериды, холестерин, фос-фолипиды, гликолипиды, стерины и воски.
Триацилглицериды - это эфиры трехатомного спирта глицерина, в норме обеспечивающие до 40% потребляемых организмом калорий.
Жирные кислоты - самые простые по строению липиды, в природе их существует свыше 200 разновидностей, более 20 из которых представлено в тканях человека (табл. 41). Они являются предшественниками простагландинов, поддерживают жидкое состояние, присущее липидам клеточных мембран в норме, предотвращают отложение холестерина в стенках кровеносных сосудов и выполняют многие другие функции. Полиненасыщенные жирные кислоты (линолевая, линоленовая, арахидоновая) относятся
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ 293
1) выработка поджелудочной железой липо-литического фермента липазы;
2) поступление с желчью желчных кислот, эмульгирующих жиры и продукты их распада, активирующих панкреатическую липазу и участвующих во всасывании жирных кислот (всасывается комплекс жирных и желчных кислот);
3) захват продуктов переваривания липидов клетками слизистой оболочки тонкого кишечника;
4) превращение в стенке кишечника всосавшихся продуктов гидролиза липидов в частицы (хиломикроны) для дальнейшего транспорта их в лимфатические сосуды и далее в кровоток.
При нарушении любого из этих процессов развивается стеаторея - избыточное содержание жира в испражнениях.
Причинами нарушения переваривания и всасывания липидов являются:
1. Дефицит или низкая активность панкреатической липазы (поражение поджелудочной железы), что приводит к нарушению расщепления жиров.
2. Недостаточное поступление желчных кислот в кишечник (при гепатитах, циррозах, холециститах, обтурационной желтухе и др.) вызывает нарушение эмульгирования и расщепления жира, а также переноса продуктов его гидролиза к всасывающей поверхности эпителия кишечника.
3. Дефицит гормопов желудочно-кишечного тракта (холецистокинин, гастрин и др.), регулирующих сокращение стенок желчного пузыря, процессы эмульгирования и расщепления жиров, их транспорт через кишечную стенку.
4. Поражение эпителия тонкого кишечника различными ядами (флоридзин, монойодуксус-ная кислота) и инфекционными агентами, инак-тивирующими ферментные системы ресинтеза триацилглицеридов эпителия тонкого кишечника, а также процессы фосфорилирования и де-фосфорилирования в стенке кишечника.
5. Авитаминозы А, В, С.
6. Избыточное потребление с пищей ионов Саг+ и Mg2+, что приводит к образованию нерастворимых в воде солей жирных кислот (мыла).
7. Дефицит холина в пище или недостаточное его образование из метионина при малобелковом питании тормозит реабсорбцию липидов.
8. Изменение деятельности нервной и эндокринной систем: перерезка блуждающего нерва
ослабляет всасывание жиров из кишечника, аналогично действует наркоз; АКТГ и тироксин усиливают всасывание жира. При недостатке гормонов коры надпочечников или избытке адреналина всасывание жира замедляется.
9. Усиленная перистальтика кишечника и
диарея препятствуют реабсорбции большей час
ти жира.
10. Нарушение метаболизма липидов в энте-
роцитах с образованием аномальных белково-
липидных комплексов ухудшает всасывание
жира и вызывает образование жировых скопле
ний в стенке тонкого кишечника и в мелких
лимфатических протоках, что блокирует отток
лимфы.
Дефицит липидов в организме может быть связан не только с нарушением их всасывания в кишечнике, но и с усилением их выведения. Организм может терять липиды с мочой (липи-дурия), что наблюдается при липоидном нефрозе. Возможны потеря липидов сальными железами (экзема, угревая сыпь) и выход липидов из депо при травматизации больших участков жировой ткани и костного мозга.
Недостаток липидов в организме может привести:
1) к развитию гиповитаминозов (снижение содержания жирорастворимых витаминов A, D, Е, К);
2) к возникновению дефицита незаменимых полиненасыщенных жирных кислот с последующим нарушением синтеза биологически активных веществ (лейкотриены, простагландины и др.). Это, как правило, сопровождается выпадением волос, воспалительным поражением кожи, возникновением некротических очагов и экзематозных явлений, поражением почек, потерей способности к размножению;
3) к развитию истощения.
11.5.2. Гиперлипемия
Гиперлипемия является одним из показателей нарушения жирового обмена и характеризуется увеличением содержания липидов в крови.
Липиды поступают в лимфу, а затем в кровь из кишечника в виде самых крупных липопро-теидов - хиломикронов, из печени в кровь выходят липопротеиды очень низкой плотности. При липолизе из подкожной жировой клетчат-
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ

|

|
| Жировая ткань |
| Желудочно-кишечный тракт |
| нэжк Липопротеидпипаза ------ И—► Избыток липопротеидов |
| Сосуд |
| Жирная кислота |
Рис. 89. Причины гиперлипемии (схема): / - усиленное поступление в кровь хиломикронов и жирных кислот из кишечника; 2 - усиленное поступление в кровь липопротеидов из печени; 3 - усиленное поступление в кровь НЭЖК из жировой ткани; 4 - низкая активность липопротеидлипа-зы; 5-6 - задержка поступления жирных кислот из
крови в жировую ткань и мышцы; 7 - усиленное расщепление комплекса альбумина с жирной кислотой; 8 - гипоальбуминемия и недостаточное образованно комплекса альбумина с жирной кислотой. Заштрихованные столбики - место нарушения процесса
ки, легких, костного мозга освобождаются не-этерифицированные жирные кислоты (НЭЖК).
Уровень липидов в плазме крови в норме не превышает 1-2 г/л.
Гиперлипемия может быть алиментарной, транспортной и ретенционной.
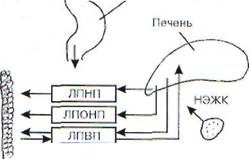
Алиментарная гиперлипемия - временное увеличение уровня хиломикронов в крови, вызванное приемом жирной пищи или проведением пробы с липидной нагрузкой. Она легко устраняется с помощью возросшей функциональной активности гепатоцитов, утилизирующих хило-микроны. Возможно также усиление депонирования липидов в жировой ткани (рис. 89).
Транспортная гиперлипемия обусловлена либо усиленной мобилизацией из депо в виде неэтерифицированных жирных кислот при голодании, стрессе, сахарном диабете, либо нару-
шением метаболизма циркулирующих в крови липопротеидов при различных формах семейной гиперлипемии. Усилению мобилизации липидов из жировой ткани, костного мозга способствуют соматотропный и кортикотропный гормоны гипофиза, а также глюкагон, тироксин и адреналин, которые активируют тканевую липазу через аденилатциклазную систему.
Из печени липопротеиды (комплекс липидов с белками) поступают в кровь. Сами липиды гидрофобии и поэтому не образуют суспензии в плазме крови. Гидрофильность им обеспечивают белки.
Мобилизация жира из легких, приводящая к гиперлипемии, возникает также при длительной гипервентиляции легких, например у пловцов и профессиональных певцов.
Ретенционная гиперлипемия (от лат. retentio
- задерживать) развивается в результате задер
жки перехода нейтральных жиров из крови в
ткани. Возникает при атеросклерозе, ишемичес-
кой болезни сердца, нефрозе, сахарном диабете,
при механической желтухе, поступлении боль
шого количества NaCl (ингибирует липопротеи-
новую липазу). В патогенезе этого вида гипер
липемии большое значение имеют следующие
факторы:
1. Снижение уровня гепарина, активирующего
фактор просветления (липопротеиновая липаза),
- при нефрозе, механической желтухе, атероск
лерозе.
2. Уменьшение содержания альбуминов в крови (осуществляют транспорт НЭЖК в клетки различных органов) - при нефротическом синдроме, заболеваниях печени и др.
3. Присутствие в сыворотке ингибитора ли-попротеиновой липазы - при нефротическом синдроме.
4. Снижение активности липокаина, активирующего поступление в кровь липопротеиновой липазы - при сахарном диабете.
11.5.3. Нарушение обмена липопротеидов (гиперлипопротеидемии и дислипопротеидемии)
Всосавшиеся в кровь неполярные липидные молекулы циркулируют в крови и лимфе в комплексе с полярными соединениями (белками). Существует большой спектр частиц, несколько
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
| Хиломикроны Ш Желудок |
 Жировая клетка
Жировая клетка
Эндотелий сосуда
Рис. 90. Обмен липидов (схема)
отличающихся по размерам, плотности и составу. Среди них выделены 4 основные группы липопротеидов (ЛП).
1. Липопротеиды высокой плотности (ЛПВП, или а-ЛП). В состав ЛПВП входят 40-55% белка (процент от общей массы частицы), 27-30% фосфолипидов, 3-8% триглицеридов, 2-3% свободного холестерина, 14-20% эфиров холестерина. Они синтезируются паренхимой печени, в стенке тонкого кишечника и всегда присутствуют в плазме крови здоровых людей. Выполняют транспортную функцию, переводя избыток холестерина с поверхности сосудов в печень и выводя его излишек из клеток эндотелия (рис. 90).
2. Липопротеиды очень низкой плотности (ЛПОНП, или пре-Р-ЛП). Представляют очень неоднородный класс частиц с различным содержанием компонентов: 8-12% - белок, 10-12% -свободный холестерин, 18-20% - фосфолипиды, 3-6% - эфиры холестерина, около 50% - тригли-цериды. Они образуются в основном в гепатоци-тах и в меньшем количестве - в слизистой кишечника, являются главной транспортной фор-
мой эндогенных триглицеридов. В плазме крови происходит трансформация ЛПОНП в Р-ЛП (при участии ферментов липопротеидлипазы и лецитин-холестеринацилтрансферазы - ЛХАТ крови). В ходе их катаболизма размеры частиц уменьшаются, меняется их состав (теряются триглицериды и возрастает относительный процент холестерина).
3. Липопротеиды низкой плотности (ЛПНП, или р-ЛП) имеют следующий состав: 24-31% -свободный холестерин, 16-28% - этерифициро-ванный холестерин, 7-11% - триглицериды, около 30% - фосфолипиды, 20-25% - белок. Они образуются в плазме из ЛПОНП и являются самой атерогенной фракцией липопротеидов у человека.
4. Хиломикроны (ХМ) - самые крупные ли-попротеидные частицы, поступающие в кровь из лимфы и представляющие собой транспортную форму пищевых жиров (экзогенных триглицеридов). В их составе находятся: 3-8% фосфолипидов, 2-4% эфиров холестерина, около 2% свободного холестерина, 1-2% белка и 86-94% триглицеридов. Хиломикроны образуются в стенке кишечника в процессе всасывания экзогенных триглицеридов и холестерина, проникают в лимфу, а оттуда в кровеносные сосуды. В плазме крови они расщепляются под действием липопротеидлипазы и теряют значительное количество триглицеридов (образуются СЖК и глицерин). Для ткани легких катаболизм ХМ особенно важен, поскольку играет ключевую роль в обеспечении высокой активности альвеолярных макрофагов и необходим для синтеза фосфолипидов сурфактанта (рис.91). В связи с этим при заболеваниях легких положительный эффект дает жировая диета. Следует отметить, что плазма крови здоровых людей натощак (через 12-14 ч после приема пищи) не содержит ХМ.
При ряде заболеваний липопротеидный спектр

|

|
Вдох
Выдох
Ателектаз
Расправленная альвеола на вдохе
Сжавшаяся альвеола на выдохе
Спавшаяся альвеола при нарушении сурфактанта
Рис. 91. Роль сурфактанта в предупреждении ателектаза
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
сыворотки меняется и возникают гипер- или гипо-(а)липопротеидемии. При этом наблюдаются увеличение или, наоборот, снижение содержания, вплоть до полного отсутствия одного или нескольких классов липопротеидов в крови, а также появление их определенных форм (дис-липопротеидемии).
Различают 5 типов гиперлипопротеидемий (ГЛП):
I. Гиперхиломикронемия - характеризуется
высоким содержанием хиломикронов в плазме
натощак. Проявляется ксантоматозом - это от
ложение холестерина и его эфиров в купферовс-
ких клетках печени, гистиоцитах подкожной
клетчатки и сухожилиях с последующим разра
станием соединительной ткани в виде бляшек и
узлов желтоватого цвета (рис. 92).
У больных развивается гепатоспленомегалия, наблюдаются тромбоз и микронекрозы поджелудочной железы с последующим формированием хронического панкреатита, абдоминальные колики после принятия жирной пищи. На коже видны ксантомы в виде желтоватых папул. Заболевание может быть вызвано наследственным аутосомно-рецессивным дефектом липопротеино-вой липазы либо аутоиммунными заболеваниями соединительной ткани (при системной красной волчанке образуются антитела против гли-козаминогликанов, что нарушает процесс гепариновой активации липопротеидлипазы).
II. Гипер-р-липопротеидемия делится на 2
типа:
Па - увеличение содержания в крови Р-ЛП при нормальном уровне пре-р-ЛП;
Пб - увеличение содержания р-ЛП и пре-Р-ЛП.
Для заболевания характерен выраженный ксантоматоз век, кожи, роговицы, развитие ишемической болезни сердца с инфарктом миокарда в очень ранНем возрасте, атеросклероти-ческие поражения сосудов у детей. Предполагается, что в основе заболевания лежит аутосом-но-доминантный дефект рецепторов ЛПНП (Па), либо нарушение взаимодействия рецепторов на клеточных мембранах с ЛПОНП и ЛПНП, либо дефект липопротеидлипазы (Пб).
III. «Флотирующая» гиперлипопротеиде-мия, или дис-Р-липопротеидемия. В основе заболевания лежит наследственно обусловленное нарушение синтеза апопротеина Е (белок, входящий в состав ХМ и ЛПОНП). Заболевание характеризуется появлением в сыворотке фло-
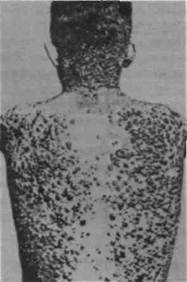
Рис. 92. Ксантоматоз кожи (по H.W. Siemens, 1921)
тирующих р-ЛП, которые называются промежуточными. Они обогащены холестерином, а содержание триглицеридов в них может быть снижено. Образуются эти частицы при нарушении катаболизма ЛПОНП и ХМ. Встречаются также приобретенные формы заболевания при гипотиреозе, танжерской болезни, некоторых аутоиммунных гаммапатиях.
Этот вид ГЛП сопровождается ранними ате-росклеротическими проявлениями (после 20 лет), развитием ИБС, ишемической энцефалопатии вплоть до инсультов, ксантоматозом, ожирением.
IV. Гипер-пре-р-липопротеидемия. Заболевание может быть наследственно обусловленным (аутосомно-доминантное) или приобретенным (при алкоголизме, остром гепатите, акромегалии, диабете и др.). Патогенез до конца не выяснен. Для этого типа ГЛП характерно нарастание уровня триглицеридов и ЛПОНП в крови. Содержание ЛПНП и ЛПВП варьирует от нормального до значительно сниженного. У больных развиваются ожирение и сахарный диабет, появляются ксантомы, возможны атеросклеротическое поражение сосудов нижних конечностей, липи-доз сетчатки и ухудшение зрения, проявления ишемической болезни сердца.
V. Гипер-пре-р-липопротеидемия и хиломикронемия. При этом заболевании в крови увеличивается содержание ХМ и ЛПОНП и снижается уровень ЛПНП и ЛПВП. У больных отме-
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
чаются гепато- и спленомегалия, ожирение, снижение толерантности к глюкозе, поражение миокарда. После приема жирной пищи могут наблюдаться внезапные приступы абдоминальной колики, ксантоматоз и атеросклероз слабо выражены.
В патогенезе первичного заболевания главную роль играет наследственно обусловленное отсутствие кофактора липопротеидлипазы - апопротеина СП (аутосомно-рецессивное наследование), в результате два основных субстрата воздействия этого фермента накапливаются в крови.
Фенокопия болезни развивается при алкоголизме, гликогенозе Гирке и некоторых других заболеваниях печени.
Гипо-(а)липопротеидемии (относительно редкие аномалии спектра ЛП)
1. А-р-липопротеидемия. В основе заболевания лежит аутосомно-доминантный дефект синтеза апопротеина В (белковой части липопроте-идов), что приводит к аномалии строения хило-микронов, снижению содержания или полному отсутствию в плазме ЛПОНП и ЛПНП. Клинические проявления связаны с нарушением всасывания в кишечнике жиров и углеводов, гемолитической анемией, дегенерацией бокового и заднего канатиков спинного мозга, пигментной ретинопатией. Нарушение всасывания жиров проявляется сразу после рождения плохим аппетитом, рвотой, обильными испражнениями, стеатореей, развитием гипотрофии. Примерно у трети больных развивается умственная отсталость. С возрастом усиливаются неврологические расстройства, появляются скелетные деформации, сердечные аритмии, ухудшается зрение. В патогенезе заболевания решающее значение имеет снижение содержания холестерина в клеточных мембранах и потеря жирорастворимых витаминов, особенно витамина Е, что ведет к утрате антиоксидантной защиты мембран.
2. Танжерская (тэнжирская) болезнь. В основе заболевания лежит аутосомно-рецессивное нарушение синтеза апопротеина А, что, в свою очередь, нарушает продукцию ЛПВП. У больных нарушен транспорт эфиров холестерина, в результате эфиры захватываются макрофагами и откладываются в клетках ретикуло-эндотели-альной системы селезенки, печени, лимфоидных
органов. Наблюдается лимфаденопатия, гепато-спленомегалия, неврологические нарушения -слабость, парестезии, снижение сухожильных рефлексов. Одним из ярких признаков заболевания является оранжево-желтый цвет увеличенных миндалин.
Существуют и другие формы гиполипопроте-идемий: церебросухожильный ксантоматоз (наследственный дефект синтеза желчных кислот из холестерина), болезнь Вальмана (аутосомно-рецессивный дефицит холинэстеразы), гипоаль-фалипопротеинемия (генетически детерминированное нарушение продукции апопротеина А и С) и др. Большинство из них связано с наследственной патологией синтеза белковой части липопротеидов либо с нарушением метаболизма холестерина.
11.5.4. Нарушение депонирования жиров (ожирение и жировая инфильтрация печени)
Ожирение - избыточное отложение жира в жировой ткани. Считается, что человек страдает ожирением, если его масса превышает нормальную более чем на 20% и продолжает увеличиваться далее. Этим недугом страдает более трети взрослого населения России. В 1998 г. ВОЗ признала ожирение хроническим заболеванием. За последнее десятилетие число таких больных в мире увеличилось почти в два раза и по оценке специалистов в 2025 г. их количество составит 300 млн человек. Ситуация тем более сложная, что с каждым годом увеличивается число молодых людей, страдающих ожирением, снижается общая продолжительность жизни населения земного шара в связи с тяжелыми заболеваниями, сопутствующими ожирению (сахарный диабет, гипертония, атеросклероз) (табл. 42).
У тучных людей старше 50 лет смертность увеличивается на 50% по сравнению с лицами, не имеющими ожирения.
Классификация ожирения
Ожирение может возникать как самостоятельное заболевание - в этом случае говорят о первичном ожирении. Вторичное ожирение - это синдром, возникающий вследствие гормональных или других расстройств в организме.
Первичное ожирение возникает при нарушении гормональной связи между жировой тканью
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
Заболевания, сопутствующие ожирению (по С. Бутровой, 2001)
Таблица -I 2
| Метаболические заболевания | ДиабетII типа,нарушенная толерантность к глюкозе, гиперинсулинемия Дислипидемия(ТГТ, ЛПВП-1, ЛПНПТ), усиленная окисляемость липопро-теинов, холецистолитиаз, гиперурикемия, жировая дистрофия печени |
| Сердечно-сосудистые заболевания | Артериальнаягипертония, ИБС,гипертрофия левого желудочка, сердечная недостаточность, венозная недостаточность |
| Новообразования | Увеличение риска развития новообразований,гормонзависимые карциномы (эндометрия, шейки матки, яичников, молочной железы, простаты), негормонзависимые карциномы (толстой кишки, прямой кишки, поджелудочной железы, печени, почек, желчного пузыря) |
| Нарушения свертываемости крови | Гиперфибриногенемия, увеличение концентрации ингибитора активатора плазминогена |
| Нарушения функции дыхательной системы | Апноэ (остановка дыхания) во сне, пиквикский синдром |
| Заболевания опорно-двигательного аппарата и кожи | Артроз коленного сустава и другие дегенеративные заболевания суставов, интертригинозный дерматит |
| Сексуальные расстройства | Нарушение менструального цикла, снижение фертильности, потеря либидо |
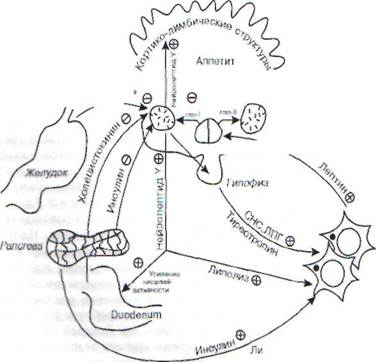
и гипоталамусом. Его главная черта - абсолютная или относительная лептиновая недостаточность. В 1994 г. был обнаружен пептидный гормон адипоцитов (клетки жировой ткани) лептин. Клетки генерируют его при накоплении жира. Лептин действует на вентролатеральные ядра гипоталамуса и вызывает чувство сытости, вен-тромедиальные ядра (центр голода) при этом тормозятся. Существует тесная связь между продукцией лептина и выработкой гормонов желудочно-кишечным трактом, нейромедиаторов, инсулина, глюкокортикоидов и др. (рис. 93).
Вторичное ожирение - синдром, возникающий при нарушении соотношения между процессами липолиза и липогенеза, носит симптоматический характер и порождается различными эндокринными расстройствами (гиперкорти-золизм, гиперинсулинизм, гипогонадизм, опухоли мозга, нарушения мозгового кровообращения и пр.).
По степени увеличения массы тела различают ожирение I степени (масса тела увеличена на 30%); II степени (на 30-50%) и III степени (более чем на 50%).
Одним из наиболее распространенных показателей для оценки степени ожирения является индекс массы тела (ИМТ), рассчитываемый следующим образом:
Масса тела в кг
ИМТ = --------------------------------------.
(Рост в м)2
Больные с ИМТ 30 кг/м2 и более, а также пациенты с ИМТ 27 кг/м2 или более, ожирение которых связано с такими факторами риска, как диабет II типа или дислипидемия, подлежат обязательному лечению (табл. 43).
Наиболее простым методом определения необходимости регулирования массы тела является измерение окружности талии. В идеале окружность талии не должна превышать 94 см у мужчин и 80 см у женщин. Если окружность талии у мужчин достигает 102 см, а у женщин -88 см, возникает серьезная угроза увеличения риска заболевания.
По особенностям морфологии жировой ткани выделяют гипертрофическое и гиперпластическое ожирение.
Гипертрофическое ожирение связано с увеличением размеров адипоцитов (это лабильный фактор, зависимый от питания), чаще встречается в зрелом возрасте. При этом виде ожирения масса тела может увеличиваться в 3-3,3 раза.
Гиперпластическое ожирение сопровождается увеличением количества адипоцитов. Начинается, как правило, в детском возрасте, так как дифференцировка фибробластических клеток-предшественниц в новые адипоциты во взрослом организме - явление довольно редкое (это происходит в период внутриутробного развития и в раннем грудном возрасте). Данный вид ожирения в большей степени зависит от наследственности. Избыток массы тела при гиперпластичес-
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ 299
Классификация избыточной массы у взрослых в зависимости от ИМТ (подготовлено в соответствии с докладом ВОЗ 1998)
Таблица 43
| Классификация | ИМТ, кг/м2 | Вероятность сопутствующего заболевания |
| Недостаточная масса | < 18,5 | Низкая (но риск других клинических проблем увеличивается) |
| Нормальный диапазон | 18,5- 24,9 | Средняя |
| Избыточная масса Предожирение Ожирение класс 1 Ожирение класс II Ожирение класс III | >25,0 25,0-29,9 30,0-34,9 35,0-39,9 >40,0 | Увеличена Умеренно увеличена Значительно увеличена Очень увеличена |
ком ожирении может достигать гигантских величин (до 1000%). Следует отметить, что в подростковый и преклимактерический периоды повышается пролиферативная активность преади-поцитов. Кроме того, их деление индуцируют избыточная калорийность пищи, сахарный диабет или переедание у беременных. В этих случаях гиперпластическое ожирение развивается у взрослых.
По патогенезу различают алиментарное, метаболическое и энергетическое ожирение.
Алиментарное ожирение - наблюдается при
чрезмерном потреблении пищи, что может быть обусловлено:
а) нарушением деятельности гипоталамичес-
кого пищевого центра (длительное возбуждение
вентролатеральных ядер) в результате травм,
кровоизлияний в диэнцефальную область, вос
паления (по этиологии это гипоталамическое
ожирение);
б) частой афферентной импульсацией при воз
буждении вкусовых рецепторов;
в) переходом от активного к малоподвижно
му образу жизни. При этом в некоторых случа-
| © Гипоталамус Вентролатеральные ядра (центр голода) Вентромедиальные ядра (центр насыщения) |
| погенеэ Продукция лептина |
 300
300
Рис. 93. Липостат и механизмы первичного ожирения (по А.Ш. Зайчику, 1999). Липостат - система, контролирующая постоянство массы тела. Ее функционирование обеспечивается взаимодействием между гипоталамусом, жировой тканью и желудочно-кишечным трактом. Холецистокинин - ингибитор голода. Нейтропептид V стимулирует поиск пищи, продукцию инсулина и липоге-нез. Его выработка уменьшается
| Адипоциты |
под влиянием лептина. ЛПГ (липотропный гормон гипофиза), СНС (симпатическая нервная система) и тиреотропин стимулируют липолиз. Лептин вызывает чувство насыщения (действуя на рецепторы вентромеди-альных ядер) и оказывает тормозящее влияние на центр голода через выработку ГПП I (глюкагоноподобный пептид)
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
ях сохраняется высокий уровень возбудимости пищевого центра (характерный для лиц физического труда или спортсменов), что приводит к систематическому перееданию;
г) чрезмерным растяжением стенок желудка
при его переполнении. Это снижает чувствитель
ность нервных окончаний слизистой оболочки,
и тормозящие импульсы передаются в пищевой
центр только при очень большом скоплении
пищи в желудке. В результате переедание ста
новится постоянным и возникает ожирение;
д) пожилым возрастом, что объясняется не
соответствием между прежним уровнем возбу
димости центра голода и меньшими энергозат
ратами (после £5 лет основной обмен снижается
в каждые ткосзгекукяцдо ^ л«ч щдалеръо ч\а
7,5%). Интересно отметить, что в глубокой ста
рости часто развивается исхудание, поскольку
угнетается активность пищевого центра и сни
жается переход углеводов в жиры.
Метаболическое ожирение обусловлено повышенным синтезом жира из углеводов. В обычных условиях до 30% поступающей в организм глюкозы под действием инсулина превращается в жир. При гиперфункции инсулярного аппарата этот процент возрастает. Аналогичное изменение метаболизма наблюдается при повышенной продукции пролактина (гормона передней доли гипофиза), глюкокортикоидов.
Энергетическое ожирение развивается при недостаточном использовании жиров в качестве источника энергии. Наблюдается при гиподинамии в сочетании с хорошим аппетитом, при снижении тонуса симпатической нервной системы и недостаточной продукции жиромобилизующих гормонов (СТГ, тиреоидные гормоны, катехола-мины), поскольку задерживается выход жира из депо и использование его в качестве энергетического субстрата.
По этиологии ожирение классифицируют на экзогенно-конституциональное, гипоталамичес-кое, гормональное (эндокринное).
Экзогенно-конституциональное ожирение. Определенные условия способствуют реализации наследственно обусловленной предрасположенности. Длительное повышение активности «пищевого центра» ведет к повышению аппетита (гиперфагии) и ожирению (рис. 94). Привычка переедать может быть приобретена в детстве. Так, установлено, что избыточное кормление ребенка первого года жизни способствует развитию
гиперпластического ожирения, характеризующегося увеличением объема жировых клеток. В результате создаются условия для развития ожирения.
Гипоталамическое ожирение. Является следствием поражения области гипоталамуса. Причиной могут быть перенесенные травмы головного мозга, стойкая внутричерепная гипертен-зия, опухоли мозга, менингит, а также врожденные дегенеративные изменения гипоталами-ческой области (например, синдром Фрёлиха) (рис. 95).
Гормональное ожирение. Связано как с гипо-, так и с гиперфункцией желез внутренней секре- ции и наблюдается при гяггогярео&е, г/г/гофуягдг- тщ% иояоъъух. желеа, а также при гипеоинсули-низме и гиперкортицизме. В крови таких больных повышается содержание ЛПНП и ЛПОНП, НЭЖК. При гормональном ожирении рано развивается гипертриацилглицеридемия и несколько позже - гиперхолестеринемия.
Нарушению обмена липидов сопутствует изменение углеводного обмена: развивается гипергликемия, стимулирующая секрецию инсулина и предшественника инсулина - низкоактивного гормона проинсулина. В свою очередь, секрецию проинсулина и инсулина стимулируют НЭЖК, ЛПНОП, ЛПНП. Усиленный выброс глюкокортикоидов, стимулирующих глюконеогенез, также повышает уровень инсулярных гормонов в

Рис. 94. Ожирение в связи с гиперфагией и перееданием. Матиас Галлас - полководец Тридцатилетней
войны (1618-1648 гг.). Гравюра неизвестного художника XVII в. (из фонда кафедры патофизиологии СГМУ)
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
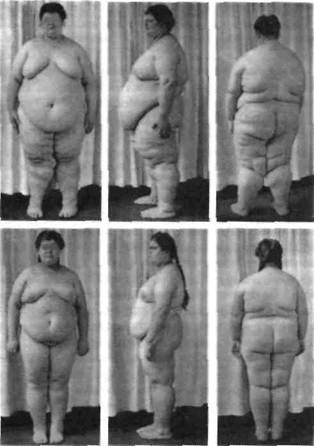
Рис. 95. Сестры с гипоталамическим типом ожирения (по D.R. Klein, 1956)
крови. Гиперинсулинизм ведет к усилению синтеза и депонирования липидов. Постепенно длительная стимуляция р-клеток инсулярного аппарата поджелудочной железы приводит к его истощению и провоцирует развитие сахарного диабета.
Последствия ожирения. При ожирении постепенно изменяется белковый обмен, который характеризуется снижением уровня общего белка крови преимущественно за счет уменьшения концентрации альбуминов, увеличением содержания фибриногена, продуктов деградации фибрина, снижением содержания гепарина. Следствием этого является нарушение транспорта НЭЖК и других липидов, повышение тромбо-генных свойств крови, снижение фибринолити-ческой активности крови, возникновение тром-боэмболических осложнений. Эти изменения являются факторами риска атеросклероза, ише-мической болезни сердца, инсульта, гипертони-
ческой болезни. Кроме этого, возникают изменения функций ЦНС, наблюдаются утомляемость, сонливость, ухудшение памяти, развивается преждевременное старение, возникают изменения во внутренних органах, например жировая инфильтрация (ожирение или жировая трансформация) печени (рис. 96).
Жировая инфильтрация печени. При нарушении расщепления и выведения жиров из клетки (гепатоцита) говорят о жировом пропитывании ее (инфильтрации). Сочетание инфильтрации с нарушением цитоплазматической структуры клетки и ее белковых компонентов называется жировой дистрофией (рис. 97).
Причинами жировой инфильтрации печени являются: сахарный диабет; ожирение; гипер-липопротеидемия; алкоголизм; отравление фосфором, мышьяком, хлороформом и другими ядами гепатотропного действия; голодание и гипо-витаминозы; инфекции и интоксикации; длительное стрессорное воздействие; недостаток в пище холина, метионина и других липотропных факторов, снижение синтеза липокаина в мелких протоках поджелудочной железы; беременность; наследственные дефекты окисления жирных кислот.
Патогенез ожирения печени связан с увеличением поступления липидов в гепатоциты и снижением их утилизации в результате торможения окисления свободных жирных кислот, нарушением образования ЛПОНП и их секреции в кровь. Например, гепатотропные яды уг-
| Желудочно-кишечный тракт |
 Жировая ткань (' Триглицериды
Жировая ткань (' Триглицериды
| Липопротеиды |
| Кетоновые тела |
| Печень |
}, Жирные к-ты
Рис. 96. Причины жировой инфильтрации печени: 1 - усиленный выход НЭЖК из жировой ткани;
2 - интенсивное длительное поступление хиломикро-
нов из кишечника в кровь, а затем в печень;
3 - задержка окисления жирных кислот в печени до
кетоновых тел; 4 - задержка выхода из печени
пре-Р-, Р-липопротеидов.
Затемненные прямоугольники - место нарушения
процесса
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
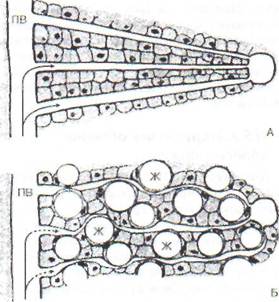
Рис. 97. Компрессия синусоидов и междольковых артерий при выраженной жировой инфильтрации печени (ПВ - портальная вена, Ж - жиры в гепато-
цитах): А - нормальная печень; Б - гепатоциты, перегруженные липидами, это может сопровождаться повышением давления в портальной вене (портальная гипертензия). При нарушении междолько-вого кровотока в центре дольки возникают гипоксия, клеточная атрофия и происходит гибель клеток
нетают окисление свободных жирных кислот (СЖК) в митохондриях печени, нарушают образование ЛПОНП, образуют активные кислородные радикалы, повреждающие гепатоциты.
Ожирение печени может заканчиваться гибелью гепатоцитов и формированием фиброза и цирроза органа. В то же время следует знать, что в принципе это процесс обратимый и в некоторых случаях он может протекать бессимптомно. Однако чаще при ожирении печени выявляются патологические печеночные пробы, гипер-кетонемия и ацидоз (в крови обнаруживаются ацетон, ацетоуксусная и р-оксимасляная кислоты), появляются признаки печеночно-клеточной недостаточности и энцефалопатии.
11.5.5. Нарушение обмена липидов и ненасыщенных жирных кислот
Жирные кислоты окисляются в процессе Р-окисления, образуя ацетил-КоА. В печени из
двух молекул ацетил-КоА образуется ацетоаце-тил-КоА, который затем взаимодействует еще с одной молекулой ацетил-КоА. Образовавшийся Р-окси-(3-метилглутарил-КоА способен под действием фермента оксиметилглутарил-КоА-лиа-зы расщепляться на ацетоацетат и ацетил-КоА. При декарбоксилировании ацетоуксусной кислоты (ацетоацетат) образуется ацетон, при ее восстановлении - D-P-оксимасляная кислота (D-р-оксибутират). Существует второй путь синтеза кетоновых тел: при отщеплении КоА от аце-тоацетил-КоА образуется свободная ацетоуксусная кислота. Процесс катализируется ферментом деацилазой, однако его активность в печени низкая, следовательно, этот путь не имеет существенного значения. При жировой инфильтрации печени отмечается избыточное поступление НЭЖК, задержка их окисления и выведения липопротеидов из печени, происходит накопление кетоновых тел в крови (гиперкетоне-мия) и в моче (кетонурия). Возможно изменение рН крови.
Гиперкетонемия со сдвигом рН крови в кислую сторону (ацидоз) возможна при угнетении цикла Кребса (цикла трикарбоновых кислот), в котором происходит окисление кетоновых тел. Это наблюдается при голодании, сахарном диабете, стрессе различной этиологии, тяжелой мышечной работе. Однако следует помнить, что в экстремальных ситуациях из кетоновых тел путем глюконеогенеза может синтезироваться глюкоза, служащая источником энергии для работы центральной нервной системы.
При метаболизме липидов образуются полиненасыщенные высшие жирные кислоты (лино-левая, линоленовая, арахидоновая). Они являются регуляторами жизненно важных функций и процессов гомеостаза: контролируют течение иммунных реакций, физиологических родов, репаративных процессов. Во время метаболизма арахидоновой кислоты по циклооксигеназ-ному пути в организме образуются простаглан-дины, простациклины, тромбоксаны, а при работе липоксигеназного метаболического пути из арахидоновой кислоты образуются лейкотриены.
Простагландины (ПГ) G2 и Н2 являются предшественниками стабильных ПГЕ, ПГ, ПГр а также простациклинов и тромбоксанов. ПГЕ оказывают иммуномодулирующее действие, расслабляют гладкие мышцы бронхов, трахеи, ЖКТ, тормозят высвобождение медиаторов из тучных
Глава 11 / ПАТОФИЗИОЛОГИЯ ТИПОВЫХ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ
клеток. ПГ02 сокращают гладкую мускулатуру бронхов и желудочно-кишечного тракта, оказывают вазодилататорное действие, снижая артериальное давление. ПГр сокращают гладкую мускулатуру, повышают проницаемость сосудов, стимулируют высвобождение медиаторов из тучных клеток.
Тромбоксаны синтезируются преимущественно в ткани мозга, селезенки, легких и в тромбоцитах, клетках воспалительной гранулемы. Они вызывают адгезию и агрегацию тромбоцитов, способствуют развитию тромбоза при ишемичес-кой болезни сердца, оказывают вазоспастичес-кое действие.
Простациклины (например, ПГ,2) синтезируются в эндотелии сосудов, сердце, ткани матки, слизистой желудка, расслабляют гладкую мускулатуру сосудов, вызывают дезагрегацию тромбоцитов, способствуют фибринолизу.
Лейкотриены (А, В, С, D) синтезируются во всех клетках крови (кроме эритроцитов), а также в адвентиции сосудов, в тучных клетках, легких. Они способствуют сокращению гладкой мускулатуры дыхательных путей и желудочно-кишечного тракта, оказывают сосудосуживающее действие (в том числе коронарных артерий). Лейкотриены запускают синтез простагландинов и простациклинов при угнетении выброса тром-боксана, что наблюдается при анафилактическом шоке. Органом-мишенью для лейкотриенов является сердце. Выделяясь в избытке, они ин-гибируют (на 60%) сократимость сердечной мышцы, уменьшая коронарный кровоток, усиливая воспалительную реакцию.
Таким образом, биологически активные вещества, являющиеся продуктами полиненасыщенных жирных кислот, в оптимальных условиях сохраняют гомеостаз организма. В случае изменения их количественного соотношения могут развиваться различные патологические реакции.
11.5.6. Нарушение обмена фосфолипидов
Описаны некоторые наследственные заболевания и патологические состояния, связанные с избыточным отложением в тканях фосфолипидов.
Так, при болезни Гоше цереброизиды откладываются в макрофагальных клетках селезенки, печени, лимфатических узлах и в костном
мозге. При болезни Нимана - Пика в клетках различных органов наблюдается отложение фос-фатида сфингомиелина. При амавротической (от греч. amauros - темный, слепой) семейной идиотии липиды отлагаются в нервных клетках, что сопровождается атрофией зрительных нервов и слабоумием.
11.5.7. Нарушение обмена холестерина
Холестерин (ХН) - производное циклопен-гана и гидратированного фенантрена. Его название происходит от греческих слов «желчь» и «твердый», поскольку впервые описан в XVIII столетии как компонент желчных камней. Общее содержание ХН в организме человека от 100 до 300 г, преобладает свободный ХН (его почти в 3 раза больше, чем эфиров холестерина). ХН может поступать в организм с пищей (яичный желток, печень, мясо, сливочное масло, сметана и сливки), существует также эндогенный синтез холестерина в печени из ацетил-КоА. Кроме того, печень - это единственный орган, где образуются эфиры ХН, поэтому снижение уровня этери-фицированного ХН служит одним из показателей недостаточности функции печени. Суточное потребление ХН варьирует от 0,2 до 0,5 г, в самом организме образуется около 1 г в день. Роль ХН огромна, он входит в состав клеточных мембран, изменяя их жидкие свойства и проницаемость, влияет на активность мембранных ферментов, может стимулировать пролиферацию способных к делению клеток (при его избытке в мембране). Холестерин является предшественником желчных кислот и стероидных гормонов: половых, глюкокортикоидов, минералокортико-идов, а также витамина D.
Выведение холестерина из организма осуществляется несколькими путями - около 0,5 г ХН в сутки превращается в желчные кислоты и теряется с желчью через кишечник, примерно такое же его количество теряется ежесуточно с фекалиями (копростерин), наконец, около 0,1 г ХН выделяют сальные железы.
В плазме крови здорового человека содержится 5,2 - 6,2 ммоль/л холестерина. Патогенными для организма являются как избыток, так и недостаток ХН.
К возникновению гиперхолестеринемии могут привести:
1. Возбуждение симпатической нервной сис-
Часть II. ТИПОВЫЕ ПАТОЛОГИЧЕСКИЕ ПРОЦЕССЫ
темы (стресс), что способствует усиленной мобилизации жира из депо и синтезу эндогенного ХН.
2. Нарушение ресинтеза жирных кислот из ацетил-КоА, что наблюдается при сахарном диабете.
3. Нарушение выведения ХН из организма при угнетении перистальтики кишечника, дис-кинезии желчных путей, при механической желтухе.
4. Эндокринные заболевания, нарушающие обмен липидов: гипотиреоз, гиперкортицизм.
5. Беременность.
6. Нефротический синдром (нарушения ли-пидного обмена и снижение содержания альбуминов).
Date: 2015-05-23; view: 715; Нарушение авторских прав