Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Метод комплексонометрии
|
|
В основе метода находятся реакции образования комплексных соединений (металло-лигандные реакции).
Метод комплексонометрии широко распространен для определения ионов Mg2+, Zn2+, Fe2+ и многих микроэлементов.
Комплексонометрия – титриметрический метод анализа, в основе которого лежит реакция взаимодействия определяемых ионов металлов с комплексонами. Комплексоны – аминополикарбоновые кислоты и их соли, способные образовывать сразу несколько связей с ионами металлов: ковалентные – Men+ с карбоксильными группами, донорно-акцепторные – Men+ с азотом аминогрупп.
Наибольшее значение из комплексонов имеет этилендиаминтетрауксусная кислота и ее двунатриевая соль (комплексон III или трилон Б – Na2H2T). Комплексоны используют в медицине для лечения лучевой болезни, свинцовых, ртутных и других отравлений металлами-токсикантами.
Взаимодействие катиона двухзарядного металла с трилоном Б можно представить схемой:
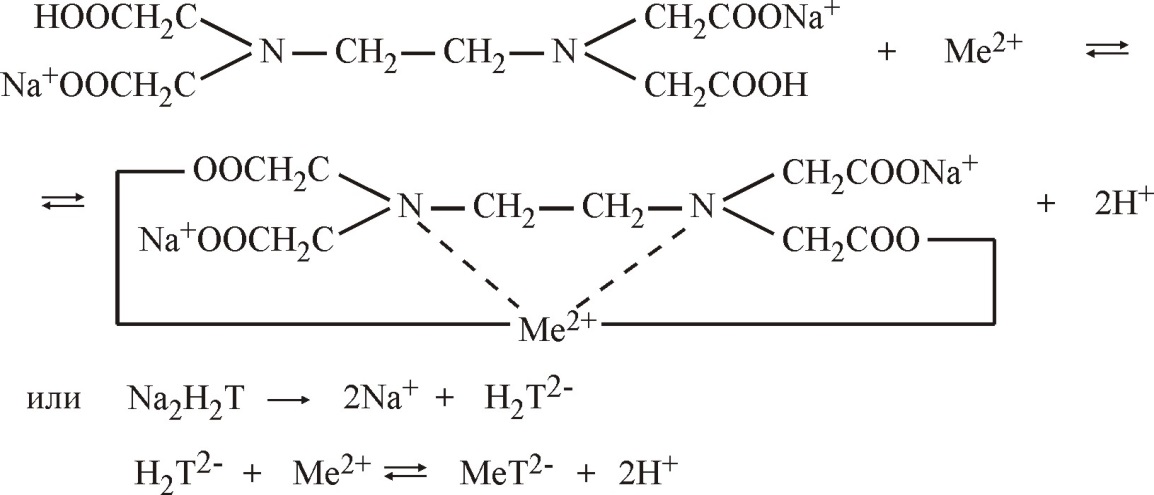
При титровании определяемых металлов, имеющих разные степени окисления, они связываются в бесцветные комплексонаты металлов, например:
Ме3+ + H2T2-  MeT- + 2H+
MeT- + 2H+
Ме4+ + H2T2- MeT0 + 2H+
Полнота протекания реакции комплексообразования увеличивается по мере связывания ионов H+ в щелочной среде. Но, в некоторых случаях, требуется создание оптимального значения pH раствора, т.к. в избытке гидроксид-ионов могут образовываться нерастворимые гидроксиды определяемых металлов. Поэтому постоянство pH во время анализа поддерживается с помощью аммиачно-амонийного буферного раствора (NH4OH – NH4Cl).
Рабочим раствором в комплексонометрии может служить раствор трилона Б. Его чаще всего готовят приблизительной концентрации, а затем стандартизируют по растворам химически чистых хлорида или сульфата магния (MgSO4∙7H2O).
Точка эквивалентности (Т.Э.) в комплексонометрии устанавливается с помощью металлохромных индикаторов. К ним относят эриохром черный Т, мурексид и др.
Метод применяют для определения жесткости воды.
3) Кислотно-основное титрование. Сущность метода и его возможности. Интервал перехода окраски кислотно-основных индикаторов. кривые титрования
Метод кислотно-основного титрования основан на реакциях взаимодействия между кислотами и основаниями, то есть на реакции нейтрализации: Н + + ОН - - Н2О
Рабочими растворами метода являются растворы сильных кислот (HCl, H2S, НNОз и др.) или сильных оснований (NaOH, КОН, Ва (ОН) 2 и др.). В зависимости от титранта метод кислотно-основного титрования подразделяют на ацидиметрию, если титрантом является раствор кислоты, и алкалиметрию, если титрантом является раствор основания.
Рабочие растворы в основном готовят как вторичные стандартные растворы, поскольку исходные для их приготовления вещества не являются стандaртными, а затем их стандартизуют по стандартным веществам или стандартным растворам. Например: растворы кислот можно стандартизовать по стандартным веществам - натрия тетраборату Na2B4О7 •10Н2О, натрия карбонату Nа2СО3 •10Н2О или по стандартным растворам NaOH, КОН; а растворы оснований - по щавелевой кислоте Н2С2О4 • 2Н2О, янтарной кислоте Н2С4Н4О4 или по стандартным растворам HCl, H2SO4, НNО3.
Точка эквивалентности и конечная точка титрования. Согласно правилу эквивалентности титрование необходимо продолжать до тех пор, пока количество прибавленного реагента не станет эквивалентным содержанию определяемого вещества. Наступающий в процессе титрования момент, когда количecтвo стандартного раствора реагента (титранта) становится теоретически строго эквивалентным количеству определяемого вещества согласно определенному уравнению химической реакции, называют точкой эквивалентности.
Точку эквивалентности устанавливают различными способами, например по изменению окраски индикатора, прибавляемого в титруемый раствор. Момент, при котором происходит наблюдаемое изменение цвета индикатора, называют конечной точкой титрования. Очень часто конечная точка титрования не совсем совпадает с точкой эквивалентности. Как правило, они отличаются друг от друга не более чем на 0,02-0,04 мл (1-2 капли) титранта. Это то количество титранта, которое необходимо для взаимодейcтвия с индикатором.
Индикаторы в методе кислотно-основного титрования
В методах кислотно-основного титрования для определения конечной точки титрования используют кислотно-основные индикаторы. Кислотно-основные индикаторы - это органические вещества, способные видимо и обратимо изменять свою окраску в растворе при изменении рН среды. Существуют различные теории индикаторов, каждая из которых по-своему объясняет поведение кислотно-основных индикаторов в кислых и щелочных средах.
Ионная теория индикаторов. В связи с тем, что кислотно-основные индикаторы представляют собой слабые кислоты или слабые основания, любой индикатор диссоциирует в растворе согласно уравнению: HInd - Н+ + Ind - бесцветный малиновый.
Окраска раствора, в котором индикатор находится в молекулярной форме (HInd), отличается от окраски раствора, в котором индикатор находится в ионной форме (Ind -). Так, моле-кулы фенолфталеина HInd бесцветны, а его анионы Ind - окрашены в малиновый цвет. Достаточно к раствору, содержащему фенолфталеин, прибавить 1-2 капли щелочи, как введенные ОН - ионы станут связывать катионы Н+ с образованием слабого электролита - молекул воды. При этом равновесие диссоциации индикатора сместится вправо, и накопление анионов Ind - вызовет окрашивание раствора в малиновый цвет.
Переход одной окраски, присущей молекулярной форме кислотно-основного индикатора, в другую, свойственную его ионной форме, происходит под влиянием Н+ или ОН--ионов, то есть зависит от рН раствора.
Хромофорная теория индикаторов. Поведение индикаторов, объясняемое ионной теорией индикаторов, дополняется хромо-форной теорией индикаторов, согласно которой изменение окраски индикаторов связано с изменением структуры их молекул, внутримолекулярной перегруппировкой, вызываемой действием Н+ или ОН--ионов. По хромофорной теории в процессе изменения рН раствора меняется строение молекул кислотно-основных индикаторов. Это явление обусловливается бензоидно-хиноидной таутомерией. При изменении рН среды раствора или при диссоциации хромофоры могут перегруппировываться. Перемена окраски у индикаторов является результатом изменений в их внутреннем строении. У одноцветных индикаторов окраска изменяется в связи с появлением или исчезновением хромофоров. У двухцветных индикаторов эти изменения обусловлены превращением одних хромофоров в другие.
Типичным одноцветным индикатором является фенол-фталеин. При рН < 8 его молекулы не содержат хиноидной груп-пировки и поэтому бесцветны. Однако при добавлении раствора щелочи к раствору фенолфталеина (рН = 8) происходит перегруппировка атомов в молекуле с образованием хиноидной группировки, которая обусловливает появление малиновой окраски раствора:
Дальнейшее увеличение рН до 13-14 вызывает другую пере-группировку, в результате чего получается трехзамещенная соль, лишённая хиноидной группировки и поэтому бесцветная:
Вследствие этого фенолфталеин обесцвечивается при действии большого избытка щелочи, например, натрия гидроксида. Типичным двухцветным индикатором является метиловый оранжевый:
(CH3) 2N N=N SO3Na
При рН = 3,2.4,3 он оранжевый, при рН? 3,1 приобретает красную, а при рН? 4,4 - желтую окраску.
Это объясняют присоединением ионов водорода кислоты к атому азота азогруппы индикатора, вследствие чего происходит смещение электронов по всей системе, сопровождающееся образованием хиноидной структуры, которая обусловливает появление красной окраски раствора. Таким образом, при действии кислот наблюдают переход желтой окраски индикатора в красную, а при действии щелочей - обратное превращение:
(CH3) 2N = =NЇN
ОН -
[ (CH3) 2N N=N SO3] + Н2 О
Цветность органических соединений, согласно хромофорной теории, обусловливается не только хиноидной структурой молекул, но и присутствием в них других хромофорных группировок (-N=N-, - N02, - NO, =С=С=, =С=О). При введении в молекулы органических веществ, содержащих хромофорные группы, ряда других групп - ауксохромов (-ОН, - Nh4, - NHR, - NHR) происходит углубление цвета окрашенного вещества.
Ионно-хромофорная теория индикаторов. Согласно дополняющим друг друга ионной и хромофорной теориям, в раст-ворах кислотно-основных индикаторов одновременно сосущест-вуют равновесия, обусловливаемые диссоциацией молекул, и равновесия, связанные с внутримолекулярными перегруппировками (ионно-хромофорная теория). Для кислотно-основных индика-торов наиболее характерными факторами, вызывающими измене-ние окраски, являются изменение соотношения количеств молеку-лярной и ионной форм индикатора, происходящее под влиянием кислот и щелочей, и появление или исчезновение хромофорных групп или же превращение одних хромофорных групп в другие.
Способность молекул различных индикаторов диссоциировать в нейтральной среде характеризуют константами диссоциации. Например, у метилового оранжевого Кa? 10 - 4, у лакмуса Кa? 10 - 8, а у фенолфталеина Кa? 10-9. Следовательно, фенолфталеин является наиболее слабой органической кислотой из этих индикаторов.
Известно, что прибавление к любому раствору любой кислоты или щелочи влечет за собой изменение концентрации ионов Н+ в нем, а следовательно, и величины рН. Перемена окраски у индика-торов также связана с изменением рН раствора. Однако каждый индикатор изменяет окраску только в определенном, характерном для него интервале значений рН. Объясняется это тем, что окраска индикатора зависит от соотношения концентраций его диссоци-ированной и недиссоциированной форм, то есть от отношения:
KHInd= [H+] [Ind-] / [HInd]
[Ind-] / [HInd] = KHInd / [H+] или [HInd] / [Ind-] = [H+] / KHInd.
Когда KHInd = [Н+], то [Ind-] / [HInd] = 1.
Если КHInd / [Н+] > 1, то в растворе превалирует диссоцииро-ванная форма индикатора, а если КHInd / [Н+] < 1, то превалирует недиссоциированная форма.
При одной и той же концентрации ионов водорода отношение КHInd / [Н+] будет тем больше, чем больше КHInd.
Для фенолфталеина КHInd = [Н+] [Ind-] / [HInd]? 10-9.
При рН = 7 [Н+] = 10 - 7, а [HInd] / [Ind-] = 10-7/10-9,
то есть при рН = 7 на каждые 100 бесцветных молекул фенолфталеина приходится лишь 1 окрашенный ион, следовательно, раствор - бесцветный. Если к раствору фенолфталеина прибавить щелочь и довести рН раствора до 8, то соотношение [HInd] / [Ind - ] = 10 - 8 /10-9 (уменьшится в 10 раз), и раствор станет бледно розовым. А при рН=9 соотношение [HInd] / [Ind-] = 10-9/10-9 = 1, то есть в растворе присутствуют равные количества бесцветных молекул индикатора и окрашенных в красный цвет ионов и раствор приобретает розовую окраску.
Таким образом, переходная окраска индикатора появляется при рН среды, равном рКHInd, но так как изменение цвета индикатора происходит постепенно, цвет недиссоциированных молекул индикатора начинает маскироваться цветом ионов задолго до достижения соотношения [HInd] / [Ind-] = 1.
Следовательно, цвет водного раствора индикатора определяется соотношением концентрации его молекулярной и ионной форм, отличающихся различной окраской, и зависит от [Н+]. Величину рН, до которой титруют раствор с данным индикатором, называют показателем титрования этого индикатора рТ.
Важнейшие индикаторы имеют следующие области перехода и показатели титрования:
Показатель титрования рТ Область перехода рН
Метиловый оранжевый…4,0…………… 3,1 - 4,4
Метиловый красный…. 5,5…………… 4,4 - 6,2
Лакмус……………… ……7,0…………… 5,0 - 8,0
Фенолфталеин……………9,0…… …….8,0 - 10,0
Кривые титрования. выбор индикатора
Кривая кислотно-основного титрования - это графическое изображение изменения рН раствора в ходе титрования.
Титрование сильной кислоты сильным основанием.
Допустим, что для титрования взяли 20 см3 раствора 0,1 моль/дм3 HCl, а в качестве титранта использовали раствор 0,1 моль/дм3 NaOH. Поскольку каждая молекула НСl дает при диссоциации один ион Н+, общая концентрация водородных ионов в 1 дм3 исходной 0,1 моль/дм3 кислоты составляет 0,1 (или 10 - 1) моль-ион. Следовательно, рН этого раствора равен 1.
Когда 90 % соляной кислоты будет оттитровано, ионов Н+ останется 10 % от первоначального количества, то есть 0,01 (или 10-2) моль-ион в 1 дм3, а рН раствора станет равен 2. При нейтрализации 99,0 % соляной кислоты рН = 3; при нейтрализации 99,9 % кислоты рН = 4 и т.д. В момент полной нейтрализации соляной кислоты титруемый раствор содержит только натрия хлорид и имеет рН = 7. Прибавление избытка натрия гидроксида ведет к увеличению рН раствора, как это показано в табл.3.1.
Результаты этих вычислений изображают графически. На оси абс-цисс откладывают избыток кислоты или щелочи в разные моменты титрования, а на оси ординат - соответствующие значения рН раствора. Получающийся график называют кривой титрования.
Ход этой кривой свидетельствует, что в конце титрования сильной кислоты сильным основанием происходит резкий скачок в измене-нии рН раствора. К моменту нейтрализации 99,9 % кислоты рН постепенно растет от 1 до 4, то есть всего на три единицы, а при переходе от 0,1 % остатка НСl к 0,1 % избытку NaOH рН раствора резко увеличивается с 4 до 10. Это означает, что добавление одной капли щелочи в конце титрования понижает концентрацию ионов Н+ с 10 - 4 до 10 - 10 моль в литре или в миллион раз.
В результате резкого изменения рН раствора от последней капли раствора основания происходит и резкое изменение окраски индикатора. При отсутствии скачка рН на кривой титрования окраска индикатора изменялась бы постепенно и определить точку эквивалентности было бы невозможно.
Титрование слабой кислоты сильным основанием
Кривую титрования слабой кислоты сильным основанием рассчитывают несколько иначе, так как при этом концентрацию ионов Н+ уже нельзя приравнивать к общей концентрации кислоты. Ее приходится вычислять с учетом константы диссоциации кислоты. Не вдаваясь в подробности вычислений, приведем кривую титрования раствора 0,1 моль/дм3 уксусной кислоты раствором 0,1 моль/дм3 NaOH (рис.3.2). Интервал скачка рН на ней значительно уже, чем в первом случае. Он простирается от рН = 7,8 (остатка кислоты в 0,1 %) до рН = 10 (?pH = 2,2 избытка щелочи в 0,1 %). Слабая уксусная кислота посылает в раствор гораздо меньше ионов Н+, чем хлороводородная. Поэтому перед началом титрования рН раствора 0,1 моль/дм3 уксусной кислоты равен 3, а не 1, как в случае с хлороводородной кислотой. В ходе титрования рН раствора уксусной кислоты все время остается выше, чем при тех же концентрациях хлороводородной кислоты. Поэтому и скачок на кривой начинается с более высокого значения рН. Заканчивается он, как и впервом случае, при рН = 10, так как титрование производят тем же раствором 0,1 моль/дм3 NaOH.
Получающийся в результате титрования натрия ацетат СН3СООNа гидролизуется с образованием некоторого избытка ионов ОН -. В связи с этим при титровании слабой кислоты сильной щелочью точка эквивалентности не совпадает с точкой нейтральности. Она лежит в щелочной области при рН = 8,9.
Для титрования слабой кислоты сильным основанием пригоден индикатор фенолфталеин. Его показатель титрования входит в пределы скачка рН на кривой и почти совпадает с точкой эквивалентности. Другие индикаторы (метиловый оранжевый, метиловый красный и даже лакмус) не могут быть использованы в этом случае, так как показатели титрования их не входят в интервал скачка.
Нейтрaлизация слабой кислоты слабым основанием
Выше было показано, что при титровании слабой кислоты сильным основанием сужается интервал скачка на кривой в области кислот-ных значений рН. И наоборот, при титровании слабого основания сильной кислотой интервал скачка уменьшается в щелочной обла-сти рН. Если попытаться титровать слабую кислоту слабым основа-нием (или наоборот), то сужение интервала скачка происходит и в кислотной, и в щелочной областях рН. Вследствие этого интервал скачка рН на кривой вовсе исчезает. На рис.3.4 показан ход изменения рН раствора при нейтрализации раствора 0,1 моль/дм3 СН3СООН раствором 0,1 моль/дм3 NН4ОН. Приведенная кривая не имеет скачка рН. Поэтому в данном случае нельзя ожидать и резкого изменения окраски индикаторов Титрование слабой кислоты слабым основанием вообще невозможно.
Выбор индикатора по продуктам реакции
В случае экспрессного анализа, когда нет возможности провести титрование стандартного образца и построить кривую титрования, чтобы максимально точно подобрать индикатор, его выбирают по продуктам реакции. Это более быстрый, но менее точный способ выбора индикатора. Как пользоваться этим методом, рассмотрим на примере количественного определения щавелевой кислоты (Ка1 = 5,62 • 10-2, Ка2 = 5,89 • 10-5):
Н2С2О4 + 2NaOH > Na2C2О4 + 2Н2О
Продуктами данной реакции являются вода (рН = 7) и гидролизующаяся соль сильного основания и слабой кислоты, гидролиз которой идет по аниону. Следовательно, в точке эквивалентности рН > 7. Значит, в данном случае подойдет индикатор, интервал перехода окраски которого лежит в щелочной среде (например, фенолфталеин).
Техника титрования
Правильное определение точки эквивалентности при титровании зависит не только от выбора индикатора, но и от порядка титрования. По методу нейтрализации титруют растворы кислот растворами оснований или наоборот. Этот порядок следует учитывать при выборе индикатора. Например, если титруют кислоту основанием и в качестве индикатора используют метиловый оранжевый (или метиловый красный), то розовая окраска индикатора от избыточной капли щелочи должна перейти в желтую. Такое изменение окраски гораздо хуже улавливается глазом, чем переход ее из желтой в розовую. Поэтому с метиловым оранжевым (или метиловым красным) рекомендуют титровать растворы оснований растворами кислот. С фенолфталeином удобнее титровать растворы кислот растворами оснований, так как при этом бесцветный раствор становится малиновым. Следует заметить, что при использовании индикаторов для фиксирования конечной точки титрования возможно появление индикаторной ошибки. Она образуется в случае несовпадения рН раствора в точке эквивалентности и рТ индикатора. Если такое несовпадение имеет место, то раствор обычно либо несколько перетитровывают, либо наоборот - недотитровывают.
Для уменьшения индикаторной ошибки титрование проводят с так называемым "свидетелем". В запасную коническую колбу (или стакан) наливают дистиллированную воду в количестве, приблизительно равном объему жидкости, получающейся в конце титрования. Прибавляют к воде столько же капель индикатора, например метилового оранжевого, сколько и к титруемому раствору, и приливают из бюретки 1-2 капли кислоты, вызывающей слабое порозовение раствора. Приготовленный таким образом "свидетель" используют в качестве образца при титровании, добиваясь, чтобы окраска анализируемого раствора и "свидетеля" была одинакова. С помощью "свидетеля" вводят также поправку в результаты титрования на прибавленный избыток кислоты, то есть из затраченного ее объема вычитают объем двух капель (? 0,04 мл), использованных на окрашивание индикатора в "свидетеле".
Наконец, правильное определение точки эквивалентности зависит от количества прибавленного индикатора. Иногда стараются прилить, побольше индикатора, полагая, что большая интенсивность окраски раствора облегчит определение точки эквивалентности. Но чем больше прибавлено индикатора, тем труднее заметить изменение окраски, так как оно будет происходить медленнее. Для установления конечной точки титрования имеет значение не столько яркость окраски раствора, сколько четкость ее изменения. Опытным путем найдено, что на 10-15 мл анализируемого раствора следует брать одну каплю раствора индикатора, а на 25 мл - не более 2 капель.
Таким образом, результат объемного определения зависит не только от выбора индикатора, но также от взятого количества его и от принятого порядка титрования.
Осадительное титрование. Классификация методов, требования к реакциям. Аргентометрия. Меркуриметрия
Методы осаждения используют для определения неорганических соединений галогенидов, цианидов, тиоцианатов, растворимых солей серебра, среди которых NaCl, КВг, КI, AgNО3 и др. входят в состав лекарственных форм. Эти методы также применяют для определения лекарственных препаратов: димедрола гидрохлорида, бромкамфоры, новокаина гидрохлорида и других. Титриметрические методы осаждения основаны на применении при титровании реакций, сопровождающихся образованием малорастворимых соединений. От гравиметрического метода осаждения они отличаются тем, что при титровании к определяемому веществу прибавляют эквивалентное количество осадителя в виде стандарт-ного раствора. Содержание определяемого компонента (вещества) рассчитывают по величине объема титранта, израсходованного на осаждение определяемого вещества. В аналитической химии известно много реакций, которые сопровождаются образованием малорастворимых соединений. Из них в количественном анализе могут быть использованы только те, которые отвечают следующим требованиям:
1. Реакция между определяемым веществом и стандартным раствором (титрантом) должна протекать в условиях, обеспечивающих образование осадка с минимальной растворимостью (S? 10 - 5 моль/дм3).
2. Реакция образования осадка должна протекать быстро, количественно, стехиометрично. При этом не должно наблюдаться образование пересыщенных растворов.
3. Должна быть возможность выбора индикатора для фиксирования конечной точки титрования.
4. Явления адсорбции и соосаждения не должны влиять на результаты определения.
Методы осаждения дают возможность количественно определять соединения, анионы которых образуют осадки с катионами:
серебра Аg+ + Вr - - AgВrv
ртути Hg22+ + 2Сl - - Hg2Cl2v
бария Ва2+ + SO42 - - BaSO4v
свинца Pb2+ + CrO42 - - PcrO4v
цинка 3Zn22+ + 2К+ + 2 [Fe (CN) 6] 22 - - Zn3К2 [Fе (СN) 6] 2v
Наиболее широко применяют методы, основанные на реакциях осаждения малорастворимых солей серебра:
Аg+ + Наl - - AgНаlv
где Наl - являются Сl, Br -, I - и др. Эти методы объединены в раздел титриметрического анализа, называемый аргентометрией.
В титриметрии применяют также метод меркурометрии, основанный на осаждении малорастворимых солей ртути (I), таких как Hg2C12, Hg2Br2, Hg2I2
Hg22+ + 2Сl - - Hg2Cl2v
Аргентометрия.
Аргентометрический титриметрический метод анализа основан на применении в качестве осадителя стандартного раствора серебра нитрата:
Аg+ + Наl - - АgНаlv
Стандартный раствор 0,1 моль/дм3 серебра нитрата может быть приготовлен:
как первичный стандартный раствор;
вторичный стандартный раствор.
Для приготовления первичного стандартного 0,1 моль/дм3 раствора AgNО3 рассчитанную навеску химически чистой соли AgNО3 взвешивают на аналитических весах, переносят в мерную колбу, растворяют в дистиллированной воде, доводят объем раствора до метки, тщательно перемешивают и переносят в склянку из темного стекла.
При приготовлении вторичного стандартного раствора АgNО3 рассчитанную навеску соли взвешивают на технических весах, переносят через воронку в склянку из темного стекла, добавляют цилиндром необходимый объем дистиллированной воды и тщательно перемешивают. Полученный вторичный стандартный раствор AgNО3 стандартизуют по химически чистым стандартным веществам KCl или NaCl или же по их растворам.
Концентрация стандартных растворов серебра нитрата изменяется при длительном хранении. Причиной нестойкости растворов серебра нитрата является их светочувствительность, потому эти растворы следует хранить в склянках из темного стекла либо в посуде, обернутой черной бумагой или покрытой черным лаком, и в защищенном от света месте. Их концентрацию необхо-димо периодически проверять.
Способы определения конечной точки титрования
В методе аргентометрии используют как безындикаторные, так и индикаторные способы фиксирования конечной точки титрования.
Безындикаторные способы
Хлорид - ионы определяют по так называемому способу равного помутнения (метод Гей-Люссака). При этом анализируемый раствор титруют стандартным раствором серебра нитрата, конец титрования определяют путем отбора двух проб титруемого раствора в две пробирки вблизи конечной точки титрования: в одну из них прибавляют каплю стандартного раствора серебра нитрата, в другую - каплю стандартного раствора натрия хлорида такой же концентрации. В недотитрованном растворе появляется помутнение в пробирке с серебра нитратом, в перетитрованном - в пробирке с натрия хлоридом. В конечной точке титрования раствор в обеих пробирках имеет одинаковое помутнение.
Бромид - и йодид - ионы определяют безындикаторным способом просветления. Суть его состоит в том, что при добавлении к анализируемому раствору из бюретки небольшими порциями стандартного раствора серебра нитрата в начале образуется коллоидный раствор серебра бромида, а в момент эквивалентности происходят коагуляция коллоидных частиц и осаждение их в виде творожистых хлопьев, раствор при этом осветляется. Этот метод достаточно точен, но в настоящее время применяется редко.
Из современных безындикаторных методов в аргентометрии чаще всего применяется потенциометрическое определение точки эквивалентности с использованием серебряных или галогенид-селективных электродов.
Индикаторные способы
Подбор индикаторов в аргентометрии очень сложен. Для его выполнения, как и в кислотно-основном методе титрования, используют кривую титрования (рис.1), представляющую собой графическое изображение изменения концентрации определяемых ионов в конце титрования, то есть когда недотитровано 10 % определяемого вещества и когда раствор перетитрован стандартным раствором на 10 %.
На кривой титрования видно, что вначале рСl изменяется медленно и лишь вблизи точки эквивалентности - скачкообразно. Резкое изменение pCl вблизи точки эквивалентности называется скачком титрования. Скачок титрования начинается, когда недотитровано 0,1 % NaCl, и заканчивается, когда раствор перетитрован на 0,1 %. Общая величина скачка титрования -
2 ед. рСl. Кривая титрования симметрична относительно точки эквивалентности ТЭКВ.
На величину скачка титрования влияют следующие факторы:
1. Концентрация реагирующих веществ: чем выше концентрация,
тем больше скачок титрования. Для растворов 1 моль/дм3 NaCl и AgNO3скачок титрования составляет 4 ед. рСl; для раствора 0,1 моль/дм3 (рассматриваемый нами случай) - 2 ед. рСl, а при концентрации 0,01 моль/дм3 - 0,3 ед. рСl;
2. Растворимость осадка: чем ниже растворимость и чем меньше произведение растворимости (ПР), тем больше скачок титрования. Например, при Ks (AgCl) = 1,78 • 10-10, Ks (AgBr) = 5,3 • 10-13 и Ks (AgI) = 8,3 • 10-17 скачок титрования для АgСl занимает 2 ед. рСl, AgBr - 4 ед. рВ и для АgI - 8 ед. соответственно.
Выбор индикатора по кривой титрования. При выборе индикатора выбирают такой ион, который образует окрашенное соединение с ионом серебра в пределах скачка титрования, то есть при рАg = 4.6 ед. наиболее пригодным оказывается K2CrО4, т.к. Ks = 2,1 * 10 - 12 и анион CrO42 - образуют окрашенный осадок с ионами серебра при концентрации последних, отвечающих значениям в пределах скачка на кривой титрования.
Произведем расчет концентрации хромат-ионов, при которой произойдет образование осадка Ag2CrО4 в конечной точке титрования:
2Аg+ + CrO42 - - Ag2CrО4v
Концентрация ионов серебра в этот момент равна: [Аg+] = vKs (AgCl) = v1,78 * 10-10 = 1,33*10 - 5 моль/ дм3.
KS (Ag2CrО4) = [Аg+] 2 [СrО42-], отсюда [СrО42-] = Ks / [Ag+] 2.
Подставив в эту формулу значение равновесной концентрации ионов серебра, получаем:
[СrО42-] = КS / [Ag+] 2 = 2,1 • 10-12/ (1,33 • 10-5) 2 = 2,1 • 10-12/1,78 • 10-10? 1 • 10-2 моль/дм3
Таким образом, если концентрация хромат-ионов в растворе будет не менее 1•10 - 2 моль/дм3, то после полного осаждения Сl - ионов образуется кирпично-красный осадок серебра хромата, что укажет на конец титрования.
В зависимости от применяемого индикатора в аргентометрии различают следующие методы:
· метод Мора, основанный на реакции между ионами серебра и галогенид - ионами в присутствии индикатора - раствора калия хромата;
· метод Фольгарда (тиоцианатометрия), основанный на реакции между ионами серебра и тиоцианат-ионами в присутствии ионов железа (III) в качестве индикатора;
· метод Фаянса - Ходакова основан на применении адсорбционных индикаторов.
Метод мора
Титрантом метода является раствор 0,1 (или 0,05; 0,02; 0,01) моль/дм3 серебра нитрата. В качестве индикатора используют раствор 0,01 моль/дм3 калия хромата, применение которого основано на дробном осаждении. Суть метода заключается в том, что при титровании галогенид-ионов в присутствии хромат-ионов в первую очередь осаждаются галогенид-ионы:
Hal - + Ag + - AgHalv
Когда определяемые галогенид-ионы практически полностью осаждаются в виде AgHal, только тогда начинает выпадать кирпично-красный осадок Ag2СrО4:
СrО42 - + 2Ag+ - Ag2СrО4v
Это обусловлено различной растворимостью солей АgНаl и Ag2CrO4v. Рассмотрим это на примере титрования раствора 0,1 моль/дм3 КСI раствором 0,1 моль/дм3 AgNО3 в присутствии индикатора - раствора 0,01 моль/дм3 K2CrО4. В растворе АgСl величина Ks (AgCl) = 1,78 • 10 - 10 достигается при концентрации ионов Ag+, равной:
[Ag+] = Ks (AgCl) / [Cl-] = 1,78 • 10-10 / 10-1 = 1,78 • 10-9 моль/дм3.
Осаждение Ag2CrО4 с величиной Ks= 1,1 • 10-12 начнется при концентрации ионов серебра, равной:
[Ag+] = v Ks (Ag2CrO4) / 10-2 = v 1,1 • 10-12/10-2 =1,05 • 10-5
Так как Ks (AgCl) достигается при меньшей концентрации ионов серебра (1,78 • 10-9 моль/дм3), чем KS (Ag2CrО4) ([Ag+] = 1,05 • 10 - 5 моль/дм3), то первым осаждается АgСl.
По мере прибавления титранта концентрация ионов серебра в растворе возрастает, и при [Ag+] = 1,05 • 10-5 моль/дм3 наряду с AgСl образуется осадок AgCrO4. Титрование заканчивают, когда взмученный в жидкости осадок от одной капли титранта (раствора серебра нитрата) приобретает красно-оранжевую окраску (начало выпадения осадка AgCrO4).
В этот момент концентрация хлорид-ионов в растворе будет равна:
[Cl-] = KS (AgCl) / [Ag+] = 1,78 • 10-10 / 1,05 • 10-5 = 1,7 • 10-5моль/дм3.
Следовательно, в данных условиях выпадение осадка серебра хромата начинается только после практически полного осаждения Сl-ионов.
Условия титрования по методу Мора:
1. Титрование следует проводить в нейтральной или слабощелочной среде (6,5? рН? 10). Метод нельзя применять:
а) в кислой среде из-за растворяемости осадка Ag2CrО4:
2Аg2СгО4. + 2Н+ > 4Аg+ + Cr2O72 - + Н2О
б) в щелочной среде из-за разложения титранта с образованием осадка Аg2О:
2Ag+ + 2ОН - - 2AgOHv > Аg2Оv + Н2О
2. В растворе должны отсутствовать:
а) катионы Рb2+, Ва2+, Hg2+ и другие, образующие с анионами индикатора осадки хроматов:
Рb2+ + CrO42 - - РbCrO4v
б) анионы PO43-, CO32-, C2О42-, AsО43 - и другие, образующие осадки с ионами серебра:
2Ag+ + CO32 - - Ag2СОз.
3. Вблизи конечной точки титрования раствор необходимо титро-вать медленно, при энергичном перемешивании, чтобы уменьшить ошибку за счет адсорбции.
4. Нельзя титровать окрашенные растворы, так как они будут маскировать окраску AgCrO4, что затруднит фиксирование конечной точки титрования.
Метод Мора применим для определения хлоридов и бромидов, в том числе фармацевтических препаратов, в состав которых входят хлорид - и бромид - ионы.
Метод Мора нельзя использовать для определения:
йодид - и роданид-ионов из-за их сильной адсорбции на поверхности осадка;
солей галоидоводородных кислот и слабых оснований, так как в peзультате гидролиза в их растворах образуется кислая среда:
NH4+ + Н2О - NНз • Н2О + Н+
Метод Фольгарда (тиоцианатометрия, роданометрия)
Метод Фольгарда основан на титровании раствора, содержащего ионы серебра, стандартными растворами NH4NCS или KNCS:
Ag+ + NCS - - AgNCSv
Индикатором в этом методе являются ионы Fe3+. После осаждения ионов серебра в виде белого осадка AgNCS избыточная капля титранта реагирует с индикатором - раствором железоаммонийных квасцов NH4 [Fe (SO4) 2] •12Н2О с образованием растворимого красного комплекса:
Fе3+ + 3NCS - - [Fе (NСS) 3]
Ионы Fe3+ образуют с NCS - ионами окрашенные комплексы различного состава: [Fe (NCS)] 2+, [Fe (NCS) 2] + • [Fе (NСS) 6] 3 - и другие, но образование комплексов различного состава не влияет на результаты титрования, так как все комплексы окрашены.
При определении по методу Фольгарда применяют прямое и обратное титрование. В качестве стандартных растворов используют:
а) в методе прямого титрования - растворы аммония тиоцианата или калия тиоцианата;
б) в методе обратного титрования - растворы серебра нитрата и аммония или калия тиоцианата.
Приготовление раствора NH4NCS. Аммония тиоцианат не является стандapтным веществом, так как соль гигроскопична. Поэтому из нее готовят раствор требуемой концентрации - приблизительно 0,1 или 0,05 моль/дм3, а затем его стандартизуют по стандартному веществу AgNО3 или по стандартному раствору АgNО3.
Условия титрования по методу Фольгарда:
1. Титрование следует выполнять в кислой среде для предотвращения гидролиза индикатора - ионов Fe3+:
Fe3+ + Н2О - FeOH2+ + Н+
2. При титровании раствор необходимо энергично перемешивать для уменьшения ошибки за счет адсорбции ионов на поверхности осадка.
3. В анализируемом растворе должны отсутствовать:
а) соли ртути (1) и (II), реагирующие с NCS - ионами:
Hg22+ + 2NCS - - Hg2 (NCS) 2v, Hg22+ + 2NCS - - [Hg (NCS) 2]
б) окислители КвrО3, КМnO4 и другие, окисляющие NСS-ионы;
в) анионы F-, PO43-, C2О42 - и другие, образующие прочные комплексы с индикатором:
Fe3+ + 6F - - [FеF6] 3-
Определение ионов Ag+ по методу Фольгарда (прямое титрование)
Концентрацию ионов серебра определяют прямым титрованием стандартным раствором аммония тиоцианата (или калия тиоцианата) в присутствии ионов Fe3+.
Стандартный раствор аммония тиоцианата реагирует в первую очередь с ионами серебра, образуя малорастворимое соединение:
Ag+ + NCS - - AgNCS KS (AgNCS) = 1,1 • 10 - 12
В конечной точке титрования избыточная капля титранта реагирует с ионами Fе3+ и окрашивает раствор в красный цвет:
Fe3+ + 3NCS - - [Fе (NСS) 3] Кнест = 4 •10-2
Метод Фольгарда (прямое титрование) применяют для опред-ния:
а) содержания серебра в сплавах (предварительно растворив его точную навеску в азотной кислоте);
б) катионов серебра в коллоидных растворах (колларголе и протар-голе);
в) концентрации солей ртути (II).
Определение анионов по методу Фольгарда (обратное титрование)
Для определения анионов используется обратное титрование.
Суть определения состоит в том, что к анализируемому раствору прибавляют удвоенный минимальный, точно отмеренный объем (35,00 или 40,00 см3) стандартного раствора серебра нитрата (l-й титрант), который реагирует с определяемыми анионами, например хлорид-ионами:
Ag + + Сl - - AgClv
Непрореагировавший остаток серебра нитрата оттитровывают вторым стандартным раствором аммония тиоцианата в присутствии индикатораионов Fe3+:
Ag+ + NCS - - AgNCSv
в конце титрования избыточная капля раствора NH4NCS реагирует с ионами Fe3+: Fe3+ + 3NCS - - [Fе (NСS) 3] и раствор окрашивается в красный цвет.
При определении хлоридов возникает ошибка за счет нечеткого установления конечной точки титрования. Это связано с протеканием обменной реакции между осадком серебра хлорида и тиоцианат-ионами в растворе, так как осадок серебра тиоцианата менее растворим, чем осадок AgCl:
AgCl + NCS - - AgNCS + Сl-
KS (AgCl) = 1,78 •10 - 10; KS (AgNCS) = 1,1•10 - 12
Это приводит к значительному перерасходу титранта NH4NCS, и результаты определения будут завышены. Для устранения этой методической ошибки осадок АgСl отфильтровывают и в полученном фильтрате определяют избыток серебра нитрата. Этот способ усложняет работу. Чаще для устранения этой ошибки к анализируемому раствору прибавляют органический растворитель, не смешивающийся с водой (тетрахлорметан CCl4, бензол С6Н6 и др.). Определение момента эквивалентности в присутствии органических раствори-телей происходит достаточно четко. Это обусловлено тем, что органические растворители покрывают поверхность осадка, изолируют его от раствора, поэтому реакция между осадком AgCl и NCS - ионами практически не протекает. При определении бромидов ошибка подобного рода не возникает, так как произведение растворимости серебра бромида меньше, чем серебра роданида: KS (AgBr) = 5,6 • 10-13 < KS (AgNCS) = 1,1•10-12. При определении йодидов по методу Фольгарда возникает ошибка за счет протекания окислительно-восстановительной реакции:
+ е + Fe3+ - Fe2+
2е + 2I - - I2
_____________________
2Fe3+ + 2I - - 2Fе2+ + I2
Эту ошибку исключают, прибавляя индикатор в конце титрования, только после того как будет введен избыток AgNО3, и йодид-ионы свяжутся в малорастворимое соединение AgIv:
I - + Ag + - AgIv
По методу Фольгарда можно определить:
а) катионы Ag+ - прямым титрованием;
б) анионы Сl, Вr-, I, NCS - обратным титрованием.
По сравнению с методом Мора метод Фольгарда имеет ряд преимуществ:
определение Ag+, Сl, Вr-, I-, NCS - выполняют в кислой среде;
катионы Ва2+, Pb2+ и другие, мешающие определению анионов по методу Мора, не мешают их определению по Фольгарду.
Метод Фаянса-Ходакова
Метод Фаянса-Ходакова основан на прямом титровании анионов (галогенидов, цианидов, тиоцианатов) стандартным раствором серебра нитрата в присутствии адсорбционных индикаторов.
Адсорбционные индикаторы представляют собой слабые органи-ческие кислоты, диссоциирующие на ионы:
HInd - H+ + Ind -
Анионы этих кислот вблизи точки эквивалентности адсорбируются поверхностью образующихся осадков, что приводит к изменению окраски осадка и позволяет фиксировать конечную точку титрования.
В качестве адсорбционных индикаторов в аналитической практике чаще всего используют флуоресцеин, дихлорфлуоресцеин, эозин и другие соединения
При титровании, например, хлорид-ионов ионами серебра образуется осадок серебра хлорида, склонный к образованию коллоидных растворов:
Ag+ + Сl - - AgCl
Осадки с ионной кристаллической решеткой адсорбируют на своей поверхности одноименные ионы, как правило, находящиеся в избытке. До конечной точки титрования на поверхности осадка AgCl адсорбируются находящиеся в избытке хлорид-ионы, и коллоидные частицы при обретают отрицательный заряд, то есть имеют следующее строение: [mАgСl] nCl-.
Эти заряженные частицы притягивают из раствора в качестве против ионов ионы K+ (вторичный адсорбционный слой):
{ [mАgСl] nCl- (n-x) K+}x-
Поскольку коллоидные частицы имеют отрицательный заряд, то адсорбция анионов индикатора невозможна.
В конечной точке титрования коллоидные частицы теряют заряд, и выпадает осадок хлорида серебра. Первая избыточная капля раствора AgNО3 создает в растворе избыток ионов Ag+, которые адсорбируются поверхностью осадка AgCl и придают ей положительный заряд: [mАgСl] nAg+, то есть происходит изменение знака заряда частицы. В качестве противоионов теперь адсорбируются нитрат-ионы (вторичный адсорбционный слой - мицеллы): { [mАgСl] nАg+ (n-x) NO3-}x+
Нитрат-ионы NО3 - не образуют труднорастворимого соединения с ионами коллоидной частицы, поэтому легко замещаются окрашенными анионами индикатора, что приводит к изменению окраски поверхности осадка и указывает на конечную точку титрования.
Условия титрования по методу Фаянса-Ходакова:
1. Титрование следует выполнять при определенном значении рН, так как это существенно влияет на ионизацию индикатора. Титрование с флуоресцеином необходимо проводить в нейтральной или слабощелочной среде (рН = 7.10); в кислой среде ионизация флуоресцеина будет подавляться, при этом концентрация его анионов понизится настолько, что не сможет образовываться окрашенный адсорбционный слой. Дихлорфлуоресцеин - кислота более сильная, чем флуоресцеин, поэтому титрование можно проводить в слабокислой среде. Эозин более сильная кислота, поэтому его можно применять как индикатор в кислой среде при рН? 2.
2. Титрование с адсорбционным индикатором следует проводить при большой поверхности осадка. Это достигается, когда осадок присутствует в виде коллоидных частиц. С этой целью к титруемому раствору прибавляют защитные коллоиды - декстрин, крахмал и др.
3. Необходимо, чтобы ионы индикатора адсорбировались осадком значительно слабее, чем определяемые ионы, иначе ионы индикатора будут адсорбироваться значительно раньше момента эквивалентности, что приведет к заниженным результатам анализа.
Метод Фаянса - Ходакова применим для определения Сl-, Br-, I-,CN - и NСS-ионов.
Меркурометрия
Меркурометрический метод анализа основан на образовании малорастворимых солей ртути (I) с хлоридами, бромидами, йодидами:
Hg2 2+ + 2Сl - - Hg2Cl2v KS = 1,3 • 10-18;
Hg22+ + 2Br - - Hg2Br2v KS = 5,8 • 10-23;
Hg22+ + 2I - - Hg2I2v KS = 4,5 • 10-29.
Титрант метода меркурометрии - раствор 0,1 моль/дм3 ртути (I) нитрата.
Приготовление стандартного раствора Нg2 (NО3) 2. Ртути (I) нитрат не является стандартным веществом, так как соль гигроскопична, неустойчива и содержит примеси Нg2+ - ионов. Поэтому из нее готовят вторичный стандартный раствор. Рассчитанную навеску Нg2 (NО3) 2 • 2Н2О взвешивают на технических весах, переносят в мерный стакан, прибавляют раствор 2 моль/дм3 азотной кислоты и нагревают до полного растворения навески. К полученному раствору прибавляют 4-5 капель металлической ртути. Приготовленный раствор выдерживают над металлической ртутью не менее суток, что приводит к восстановлению Hg2+ - ионов:
Hg2+ + Hg > Hg22+
Только после этого полученный раствор стандартизуют по стандартным веществам - химически чистому NaCl или КСl или же по их стандартным растворам.
Концентрация раствора ртути (1) нитрата не изменяется в течение нескольких месяцев.
В методе меркурометрии в качестве индикаторов используют:
а) раствор железа (III) тиоцианата [Fе (NСS) 3];
б) 1 % -ный раствор дифенилкарбазона в 95 % -ном спирте.
При применении раствора [Fе (NСS) 3] точку конца титрования фиксируют по исчезновению красной окраски индикатора.
Изменение окраски происходит при взаимодействии одной избыточной капли титранта с раствором индикатора:
3Hg22+ + 2 [Fе (SСN) 3] - 3 [Hg2NCS) 2] + 2Fe3+
При титровании с данным индикатором необходимо проводить контрольный опыт для установления объема титранта, израсход-ованного на реакцию с индикатором. Для этого к 20-25 см3 дистиллированной воды прибавляют все реагенты в тех же количествах, что и при анализе пробы, и титруют стандартным раствором ртути (I) нитрата. Полученный объем титранта вычитают из объема, израсходованного на титрование пробы.
Точка эквивалентности (в титриметрическом анализе) — момент титрования, когда число эквивалентов добавляемого титранта эквивалентно или равно числу эквивалентов определяемого вещества в образце. В некоторых случаях наблюдают несколько точек эквивалентности, следующих одна за другой, например, при титровании многоосновных кислот или же при титровании раствора, в котором присутствует несколько определяемых ионов.
На графике кривой титрования присутствует одна или несколько точек перегиба, соответствующих точкам эквивалентности.
Точкой окончания титрования (подобна точке эквивалентности, но не то же самое) считают момент, при котором индикатор изменяет свой цвет при колориметрическом титровании.
Методы определения точки эквивалентности[править | править исходный текст]
С помощью индикаторов[править | править исходный текст]
Это вещества, изменяющие свой цвет вследствие протекания химических процессов. Кислотно-основные индикаторы, например фенолфталеин, изменяют свой цвет в зависимости от pH раствора, в котором они находятся. Редокс-индикаторы изменяют свой цвет вслед за изменением потенциала системы, используются таким образом при окислительно-восстановительном титровании. Перед началом титрования в исследуемый раствор добавляют несколько капель индикатора и начинают по каплям добавлять титрант. Как только раствор вслед за индикатором изменяет свой цвет, титрование прекращают, этот момент приблизительно и есть точка эквивалентности.
Правило выбора индикатора - при титровании используется такой индикатор, который изменяет свою окраску около точки эквивалентности, т.е. интервал перехода окраски индикатора должен по возможности совпадать со скачком титрования.
Потенциометрия[править | править исходный текст]
В данном случае используют прибор для измерения электродного потенциала раствора. При достижении точки эквивалентности потенциал рабочего электрода резко изменяется.
С помощью pH-метров[править | править исходный текст]
pH-метр по сути своей также является потенциметром, в котором используется электрод, потенциал которого зависит от содержания в растворе ионов H+, это пример использования ионоселективного электрода. Таким образом можно следить за изменением pH в течение всего процесса титрования. При достижении точки эквивалентности pH резко изменяется. Данный способ более точный по сравнению с титрованием с использованием кислотно-основных индикаторов, и может быть легко автоматизирован.
Проводимость[править | править исходный текст]
Проводимость раствора электролитов зависит от находящихся в нем ионов. Во время титрования проводимость часто значительно изменяется (Например, при кислотно-основном титровании, ионы H+ и OH− взаимодействуют, образуя нейтральную молекулу H2O, что вызывает изменение проводимости раствора). Общая проводимость раствора зависит и от других присутствующих ионов (например, противоинов), которые вносят в нее различный вклад. Он, в свою очередь, зависит от подвижности каждого иона и от общей концентрации ионов (ионной силы). В связи с этим предсказать изменение проводимости гораздо сложнее, нежели измерить ее.
Изменение цвета[править | править исходный текст]
При протекании некоторых реакций происходит изменение цвета и без добавления индикатора. Чаще всего это наблюдается при окислительно-восстановительном титровании, когда исходные вещества и продукты реакции имеют разные цвета в разных степенях окисления.
Осаждение[править | править исходный текст]
Если во время реакции образуется твердое нерастворимое вещество, то по окончании титрования образуется преципитат. Классическим примером такой реакции является образование крайне нерастворимого хлористого серебра AgCl из ионов Ag+ и Cl−. Удивительно, но это не позволяет точно определить момент окончания титрования, поэтому осадительное титрование чаще всего используют в качестве обратного титрования.
Изотермическое калориметрическое титрование[править | править исходный текст]
Используется изотермический титровальный калориметр, который по величине тепла, которое выделила или поглотила реагирующая система, определяет точку эквивалентности. Данный способ важен в биохимическом титровании, например, для определения того, какферментный субстрат связывается с ферментом.
Термометрическая титриметрия
Термометрическая титриметрия — чрезвычайно гибкая техника. Она отличается от калориметрической титриметрии тем, что теплота реакции, о которой свидетельствует падение или рост температуры, не используется для определения количества содержащегося в исследуемом образце раствора вещества. Напротив, точка эквивалентности определяется на основе области, в которой происходит изменение температуры. В зависимости от того, является реакция между титрантом и исследуемым веществом экзотермической или эндотермической, температура в течение процесса титрования будет, соответственно, возрастать или падать. Когда все исследуемое вещество прореагировало с титрантом, изменение области, в которой происходит рост или падение температуры, позволяет определить точку эквивалентности и изгиб на кривой температуры. Точно точку эквивалентности можно определить, взяв вторую производную кривой температуры: четкий пик будет указывать на точку эквивалентности.
СпектроскопияТочку эквивалентности можно определить, измеряя абсорбцию света раствором во время титровании, если известен спектр продукта, титранта или исследуемого вещества. Относительное содержание продукта реакции и исследуемого вещества позволяют определить точку эквивалентности. При этом присутствие свободного титранта (указывающее на завершение реакции) можно обнаружить при очень малых величинах.
Амперометрия
Метод, позволяющий определить точку эквивалентности по величине тока при заданном потенциале. Величина тока вследствие реакции окисления/восстановления исследуемого вещества или продукта у рабочего электрода зависит от их концентрации в растворе. Точке эквивалентности соответствует изменение величины тока. Данный метод наиболее полезен, когда необходимо уменьшить расход титранта, например, при титровании галидов ионом Ag+.
Кисло́тность желу́дочного со́ка — характеристика концентрации кислоты в желудочном соке. Измеряется в единицах рН.
Для оценки состояния органов желудочно-кишечного тракта (ЖКТ) рассматривают величину кислотности (рН) одновременно в разных отделах желудка и, более широко, одновременно в разных отделах пищевода, желудка и двенадцатиперстной кишки; изменение рН во времени; динамику изменения рН, как реакцию на стимуляторы и лекарственные препараты.
Зоны кислотопродукции и кислотонейтрализации желудка
Желудочный этап переваривания пищи происходит с помощью ферментов, важнейшим из которых является пепсин, требующих обязательно кислой среды. Однако, кислота в химусе (кашице), состоящем из частично переваренной пищи и желудочных соков, перед эвакуацией из желудка должна быть нейтрализована.
Желудок условно можно разделить на кислотообразующую (верхнюю) и кислотонейтрализующую (нижнюю) зоны, разделённые интермедиарной зоной, то есть зоной перехода от слабокислых рН (6,0-4,0) к резкокислым (рН менее 3,0) и располагающейся между телом желудка и его антральным отделом.
Кислотообразующая зона соответствует дну и телу желудка, кислотонейтрализующая — антральному отделу желудка. Так как при исследовании кислотности желудка диагностически важной является информация о процессах кислотопродукции и кислотонейтрализации, то измерение кислотности желудка должно происходить не менее, чем в двух зонах: теле желудка и антруме.
Нейтрализация кислоты в желудке производится, в основном, за счёт ионов гидрокарбонатов (HCO3-), секретируемых поверхностными клетками слизистой оболочки.
Методы исследования кислотности желудка
Существуют четыре основных метода исследования кислотности желудочного сока.
Наиболее простой — при помощи ионообменных смол («Ацидотест», «Гастротест» и др.) по степени окрашивания мочи. Метод имеет небольшую точность и, поэтому, малоинформативен. В последнее время применяется редко.
Аспирационные методы. Наиболее распространён из них метод фракционного зондирования. Содержимое желудка отсасывается при помощи резиновой трубки, а затем исследуется в лаборатории. Этот метод имеет свои достоинства, но имеет и серьёзные недостатки. В процессе отсасывания содержимое желудка, полученное из разных функциональных зон, перемешивается. К тому же сам процесс отсасывания нарушает нормальную работу желудка, искажая результаты исследования.
Метод окрашивания стенки желудка при помощи орошения её специальным красителем через канал эндоскопа во время проведения гастроскопии. Этот метод также не может обеспечить требуемую точность, визуальное определение кислотности по изменению цвета красителя дает очень приблизительные результаты.
Исследование кислотности желудочного сока с помощью многоместного ацидогастрометра.
Электрометрический метод измерения кислотности непосредственно в желудочно-кишечном тракте — внутрижелудочная рН-метрия. Это наиболее информативный и физиологичный метод. Позволяет с помощью специальных приборов — ацидогастрометров, оснащённых рН-зондами с несколькими датчиками рН, измерять кислотность одновременно в разных зонах желудочно-кишечного тракта в течение длительного времени (до 24-х часов и более). Недостатком метода является невозможность измерения общего объёма кислотопродукции желудка.
pH в желудке и соседних отделах ЖКТ[править
Максимальная теоретически возможная кислотность в желудке: 0,86 рН (соответствует кислотопродукции 160 ммоль/л).
Минимальная теоретически возможная кислотность в желудке: 8,3 рН (соответствует рН насыщенного раствора ионов HCO3-).
Нормальная кислотность в просвете тела желудка натощак: 1,5 — 2,0 рН.
Кислотность на поверхности эпителиального слоя, обращённого в просвет желудка: 1,5 — 2,0 рН.
Кислотность в глубине эпителиального слоя желудка: около 7,0 рН.
Нормальная кислотность в антральном отделе желудка: 1,3 — 7,4 рН.
Нормальная кислотность в пищеводе: 6,0 — 7,0 рН.
Нормальная кислотность в луковице двенадцатиперстной кишки: 5,6 — 7,9 рН.
Кислотность сока тонкой кишки: 7,2 — 7,5 рН; при усилении секреции достигает 8,6 рН.[2]
Кислотность сока толстой кишки: 8,5 — 9,0 рН.[2]
Лабораторное определение кислотности желудочного сока
В лаборатории кислотность желудочного сока определяют титрованием его раствором едкого натра (NaOH) с участием различных химических индикаторов, меняющих свой цвет в зависимости от кислотности среды. Разделяют понятия общей кислотности желудочного сока, свободной и связанной кислотности.
Кислотность желудочного сока выражают или в титрационных единицах (количестве мл 0,01 М раствора едкого натра, необходимого для нейтрализации кислоты в 100 мл желудочного сока) или в ммоль/л HCl на 1 л желудочного сока. Численно эти значения совпадают. Обычно при титровании используют 5 мл желудочного сока. Поэтому, после титрования, нейтрализованное количество NaOH умножают на 20.
Общая кислотность желудочного сока
Общая кислотность складывается из свободной и связанной кислотностей плюс кислотность, обусловленная органическим кислотами (молочной, уксусной, масляной и другими) в норме или при патологии.
Для определения общей кислотности к 5 мл желудочного сока добавляют одну каплю 1 % спиртового раствора фенолфталеина. Отметив уровень раствора в мерной пробирке, производят титрацию желудочного сока до появления красного окрашивания. Количество мл едкого натра, потраченного на титрование, умноженного на 20, будет равно общей кислотности в титрационных единицах или ммоль/л.
Свободная соляная кислота
Модель молекулы соляной кислоты
Свободной соляной кислотой называется соляная кислота, находящаяся в желудочном соке в виде отдельных ионов H+ и Cl—.
Для определения свободной кислотности к 5 мл желудочного сока добавляют одну каплю диметиламидоазобензола. Отметив уровень раствора в мерной пробирке, производят титрацию желудочного сока до появления оранжево-жёлтого цвета. Количество мл едкого натра, потраченного на титрование, умноженного на 20, будет равно свободной кислотности.
Связанная соляная кислота
Связанной соляной кислотой называется соляная кислота, находящаяся в желудочном соке в химически связанном с белками и в недиссоциированном виде.
Для определения связанной соляной кислоты используют индикатор ализарин. Процедура титрования аналогична описанным выше и проводится до появления фиолетового окрашивания.
4) ПЕРМАНГАНАТОМЕТРИЯ, титриметрич. метод анализа, основанный на р-циях: 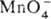 + 8H+ + 5е
+ 8H+ + 5е 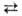 4H2O + Mn2+ и
4H2O + Mn2+ и 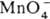 + 4H2O + Зе
+ 4H2O + Зе 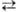 MnO2 + 4OH— (стандартные электродные потенциалы соотв. +1,52 и +0,57 В). Титрантом служит водный р-р KMnO4, к-рый в чистом виде очень устойчив и долго хранится. Однако в присут. Mn(II) происходит р-ция:
MnO2 + 4OH— (стандартные электродные потенциалы соотв. +1,52 и +0,57 В). Титрантом служит водный р-р KMnO4, к-рый в чистом виде очень устойчив и долго хранится. Однако в присут. Mn(II) происходит р-ция: 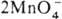 + 3Mn2+ + 2H2O
+ 3Mn2+ + 2H2O 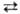 5MnO2 + 4H+, к-рая ускоряется диоксидом марганца и при понижении кислотности р-ра. Поскольку перманганат калия всегда содержит трудно удаляемые примеси, свежеприготовленный р-р KMnO4 кипятят в течение часа, фильтруют через стеклянный фильтр и хранят в темных склянках, в защищенных от прямого солнечного света местах (т.к. на свету ускоряется разложение KMnO4 на MnO2 и O2).
5MnO2 + 4H+, к-рая ускоряется диоксидом марганца и при понижении кислотности р-ра. Поскольку перманганат калия всегда содержит трудно удаляемые примеси, свежеприготовленный р-р KMnO4 кипятят в течение часа, фильтруют через стеклянный фильтр и хранят в темных склянках, в защищенных от прямого солнечного света местах (т.к. на свету ускоряется разложение KMnO4 на MnO2 и O2).
Для определения концентрации титранта используют р-ры с точно известным содержанием (стандартные р-ры) Na2C2O4, As2O3 (в присут. ICl или KIO3 в качестве катализатора), FeS04·(NH4)2SO4·6H2O и K4[Fe(CN)6]·3H2O. Конечную точку титрования в перманганатометрии устанавливают обычно визуально без индикатора по появлению или исчезновению окраски перманганат-иона (даже 2·10-6 M р-р KMnO4 имеет отчетливую розовую окраску), потенциометрически или амперометрически. При обратном титровании к исследуемому р-ру приливают р-р KMnO4, избыток к-рого от-титровывают р-ром восстановителя, напр. щавелевой к-ты.
Перманганатометрию применяют для определения Fe(II), Sb(III), Mn(II), V(IV), W(V), U(IV), Tl(I), Cr(III), H2O2, H2C2O4 и ее солей, арсенитов, гидразина и ряда орг. в-в (напр., хлоруксусной и пропионовой к-т в щелочной среде); обратным пермангана-тометрич. титрованием определяют восстановители, медленно реагирующие с KMnO4, - иодиды, цианиды, родани-ды, фосфиты.
Перманганатометрию используют также для косвенных определений. Напр., концентрации очень сильных восстановителей, окисляющихся в обычных условиях растворенным кислородом, - Cr (II), V(II), Ti(III), Nb(III), Mo(III), Cu(I), Sn(II) и др. - устанавливают после их взаимод. с ионами Fe(III) по кол-ву образовавшихся ионов Fe (II), к-рые оттитровывают р-ром KMnO4. Ионы таких металлов, как Ca, Cd, Zn, Pb(Il), Со, Ni, РЗЭ, определяют после их осаждения в видеоксалатов. К перманганатометрии часто относят обратное ферриметрич. титрование, в к-ром при определении окислителей (дихроматов, персульфатов, ванадатов, MnO2, PbO2, Pb3O4 и др.) их предварительно восстанавливают с помощью Fe(II), избыток к-рого оттитровывают р-ром KMnO4.
Каталаза
Взятие крови натощак.
Материал для исследования: плазма крови.
Каталаза - гемосодержащий фермент, осуществляющий защитную функцию в отношении перекиси водорода. Разложение перекиси водорода каталазой на молекулярный кислород и воду осуществляется в два этапа. При этом в окисленном состоянии каталаза работает и как пероксидаза, катализируя окисление спиртов или альдегидов. Кроме того, каталаза может выступать источником образования активных форм кислорода. Около 5% кислорода, образующегося в результате разложения, Н2О2 возникает в возбужденном синглетном состоянии.
Максимальное содержание каталазы обнаружено в эритроцитах, значительное количество в печени и почках. Обладает специфической антиоксидантной защитой в отношении эндотелиальных клеток.
Норма: эритроциты: 31 - 34,1 нМ Н2О2/ мг гемоглобина
плазма: 14,8 - 16,5 нМ Н2О2/ мг гемоглобина
Повышение активности: отмечается при бета-талласемии, некоторых опухолях, усилении перекисных процессов (в стадии компенсации).
Снижение активности происходит при железодефицитных анемиях, усилении пероксидации в стадии декомпенсации, а также с возрастом.
5) Йодометрия
Аналитическая химия
Йодометрия - метод окислительно-восстановительного титрования, основанный на реакциях, связанных с окислением восстановителей свободным йодом I2 или с восстановлением окислителей йодидом калия KI. Оба процесса можно выразить следующей схемой:
I2 + 2е 2 I? (2.8)
Т.к. стандартный окислительно-восстановительный потенциал пары невелик? E0(I2 /2I?) = 0,54 В, йод является относительно слабым окислителем, а ионы I? - сравнительно сильным восстановителем.
С помощью метода йодометрии можно определять:
1. Восстановители SO3-2?, S2O3-2?, NO2?, S2?, CN?, SCN? и др.:
а) путем прямого титрования анализируемого раствора раствором йода:
SO3-2? + I2 + 2Н2О = SO4-2? + 2 I? + 2H+; (2.9)
б) путем обратного титрования, Используется, если скорость взаимодействия восстановителя с йодом невелика. В этом случае к раствору восстановителя добавляют избыток титрованного раствора I2, а спустя некоторое время не вступивший в реакцию йод титруют раствором тиосульфата натрия:
2 S2O32? + I2 = S4O62- + 2 I? (2.10)
2. Окислители Fe3+, Cr2O72-, H2O2, Cl2, Br2, ClO3?, MnO4? и др.
Проводить определение, титруя раствор окислителя йодидом калия KI нельзя, т.к. невозможно фиксировать конец титрования. Поэтому пользуются методом замещения: к анализируемому раствору окислителя добавляют избыток раствора KI:2 MnO4? + 10 I? + 16 H+ = 5 I2 + 2 Mn2+ + 8 Н2О. (2.11)
В результате реакции выделяется йод в количестве, эквивалентном количеству окислителя; йод титруют раствором тиосульфата натрия по уравнению (2.10).
К достоинствам метода йодометрии можно отнести следующие:
1. Большая точность по сравнению с другими методами окислительно-восстановительного титрования.
2. Растворы йода окрашены, и титрование можно проводить без индикатора. Желтая окраска ионов I3? при отсутствии других окрашенных продуктов отчетливо видна при очень малой концентрации (5*10?5н.).
3. Йод хорошо растворяется в органических растворителях, поэтому метод широко применяется для титрования в неводных средах.
Недостатки метода, вызывающие ошибки при выполнении йодометрических определений:
1. Потери йода из-за его летучести. Поэтому титрование проводят на холоду и по возможности быстро. При необходимости оставить раствор на некоторое время для завершения реакции, его хранят под притертой пробкой.
2. Окисление ионов йода кислородом воздуха в кислой среде.
3. Йодометрическое титрование нельзя проводить в щелочной среде вследствие диспропорционирования йода.
4. Относительно медленные скорости реакций с участием йода.
5. В процессе хранения стандартные растворы йода и тиосульфата изменяю
Date: 2015-05-22; view: 16964; Нарушение авторских прав