Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Печеночная недостаточность
|
|
Все заболевания печени в зависимости от характера альтерации условно можно разделить на три большие группы: гепатоцеллюлярные (гепатиты, циррозы), холестатические, инфильтративно-опухолевые. Вторичные патологические изменения являются общими для всех групп заболеваний. Поврежденные гепатоциты нарушают нормальный отток желчи. Непроходимость или воспаление желчных путей вызывают патологические изменения в гепатоцитах независимо от причины и характера поражения.
Печеночная недостаточность – патологическое состояние, характеризующееся комплексными нарушениями обмена веществ в сочетании с поражением мозга. Печеночная недостаточность является следствием первичных (вирусный гепатит, острая и подострая дистрофия печени, возникающая под влиянием химических факторов, некоторые формы цирроза печени) и вторичных (длительно прогрессирующая, застойная желтуха, распад печеночной ткани при опухоли и другие) поражений печеночной паренхимы. В зависимости от патогенеза выделяют печеночно-клеточную (истинная) и портально-печеночную (шунтовая) недостаточность, по течению – острую и хроническую.
Острая печеночная недостаточность развивается в результате массированного некроза гепатоцитов, вызванного различными причинами (молниеносные формы острого вирусного или алкогольного гепатита, лекарственные препараты, пищевые и промышленные яды, сепсис, эндотоксикоз, развившийся после обширных резекций тонкой кишки, все виды острой гиповолемии, синдром рея).
Хроническая печеночная недостаточность развивается при прогрессировании хронических заболеваний печени (цирроз, гепатит) и опухолевых процессов, тромбозах в системе печеночной и воротной вен, портокавальных анастомозах. Прогрессирующее снижение функциональной активности гепатоцитов при морфологической их полноценности проявляется печеночной недостаточностью у пациентов с циррозом, выраженным диффузным стеатозом, страдающих хроническим алкоголизмом. В связи с огромным значением печени в обмене веществ именно ее функциональная активность, развитие острой или хронической печеночной недостаточности во многом предопределяет исход заболевания. Основные клинические признаки печеночной недостаточности – нарастающая слабость, диспепсические расстройства, желтуха, геморрагический диатез, печеночный запах изо рта, асцит. К осложнениям печеночной недостаточности в первую очередь следует отнести энцефалопатию.
Печеночная энцефалопатия, крайней степенью выраженности которой является печеночная кома, возникает вследствие повышенного поступления в головной мозг аммиака, жирных кислот с короткой цепью, меркаптанов, фенолов, ароматических аминокислот, свободных жирных кислот, свободного билирубина. Печеночная недостаточность сопровождается нарушениями в свертывающей системе крови. Возможны массивные желудочно-кишечные кровотечения, развитие ДВС-синдрома. Вторично нарушается функция сердечно-сосудистой системы, возникают отеки.
Метаболические нарушения при печеночно-клеточной недостаточности.В физиологических условиях в печени синтезируются структурные белки клеточных мембран, растворимые белки клеток, белки плазмы, гликоген, липопротеины, холестерин, желчные кислоты и другие. Активными индукторами белкового синтеза в гепатоцитах являются стероидные гормоны (эстрадиол), лекарственные препараты (фенобарбитал, преднизолон, дексаметазон). Их протективное действие заключается в подавлении активности ядерной и цитоплазматической нуклеаз и повышении уровня 45S РНК. Анаболические гормоны обладают стимулирующим влиянием на гликогенсинтетическую функцию. Использование индукторов белкового синтеза в определенной степени препятствует также и уменьшению содержания гликогена в гепатоцитах. Характер метаболических нарушений в печени зависит от гепатотоксичности повреждающего фактора, остроты и длительности патологического процесса. При поражениях печенинарушаются процессы синтеза и метаболизма белков, липидов, углеводов.
Нарушения белкового обмена характеризуется снижением белково-синтетической функции гепатоцитов в результате угнетения каталитической активности связанных с мембранами ферментов и ферментативной активности субклеточных структур. Нарушается контакт рибосом с эндоплазматическим ретикулумом вследствие редукции мембран и уменьшения их белкового компонента. Снижение белково-синтетической функции печени проявляется гипоальбуминемией, диспротеинемией (изменения α- и β-глобулинов) и уменьшеньем содержания прокоагулянтов, прежде всего, протромбинового комплекса.
Альбумин – основной белок плазмы, синтезируемый только впечени. В норме на его долю приходится 60% общего белка плазмы крови. Период полураспада альбумина колеблется в пределах 7-26 дней, и поэтому при острой печеночной недостаточности его уровень в плазме в первые 7-14 дней существенно не меняется. Альбумин выполняет в организме транспортную и антитоксическую функцию (связывание метаболитов и ксенобиотиков).
При хронической печеночной недостаточности наблюдается выраженная гипоальбуминемия. Пропорционально снижению уровня альбумина уменьшается онкотическое давление плазмы и, соответственно, объем циркулирующей крови. Гипоонкия проявляется асцитом, периферическими отеками, гипотонией. Токсикоз эндогенными и экзогенными ядами в условиях гипоальбуминемии наступает даже при их минимальном содержании в плазме.
При острой печеночной недостаточности уменьшается синтез прокоагулянтов. Это относится, в первую очередь, к факторам II и VII (группа витамин- К -зависимых прокоагулянтов). Развивается умеренная гипофибриногенемия. Каждый из факторов свертывания может снижаться в разной степени в зависимости от объема некроза печени, скорости синтеза и продолжительности жизни того или иного фактора. У 80% больных с патологией печени отмечается гипокоагуляция различной степени выраженности, причинами которой может быть не только указанное выше снижение синтетической активности, но и синтез аномальных факторов свертывания.
Для печеночной недостаточности характерно усиление катаболизма белка в мышцах и использование в качестве энергетического субстрата аминокислот с разветвленной цепью. Причем, чем интенсивнее используются эти аминокислоты в мышечной ткани, тем в больших количествах в кровь поступают ароматические и сульфатированные аминокислоты; развивается дизаминоацидемия.
Для характеристики аминокислотного спектра крови и спинномозговой жидкости определяют аминокислотное соотношение:
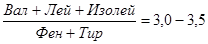
При печеночной недостаточности это соотношение снижается, при энцефалопатии оно меньше 1,0. Наиболее существенное значение в развитии печеночной энцефалопатии и комы имеет повышение содержания ароматических (триптофан, фенилаланин, тирозин) и сульфатированных (метионин, цистеин) аминокислот, а также снижение уровня аминокислот с боковой цепью (валин, лейцин, изолейцин).
Ароматические аминокислоты в высоких концентрациях угнетают ферментативную систему, превращающую тирозин в ДОФА, который преобразовывается в норадреналин и дофамин. Поэтому активация альтернативных путей метаболизма ароматических аминокислот сопровождается образованием в нервной системе ложных нейротрансмиттеров.
Триптофан. Деградация триптофана в основном происходит в печени. При печеночной недостаточности гидроксилирование триптофана снижается, нарастает его содержание в крови, и он больших количествах попадает в мозг. В результате расщепления триптофана в ткани мозга образуются аминные метаболиты – серотонин и триптамин, являющиеся в таких концентрациях более токсичными, чем сама аминокислота. Серотонин накапливается в избыточном количестве в свойственных ему областях мозга. В нейронах, где функционируют другие трансмиттеры, серотонин вытесняет естественные передатчики и выступает в роли ложного нейротрансмиттера. В определенной степени описанные процессы способствуют развитию энцефалопатии. Продуктами внутрикишечного расщепления триптофана являются индол и скатол. При снижении антитоксической функции печени они в чрезмерных количествах попадают в головной мозг и оказывают токсическое действие на клетки ЦНС. Триптофан снижает токсический порог некоторых веществ, в первую очередь, аммиака.
Метионин расщепляется в кишечнике до аммиака и меркаптанов (этантиол, метантиол). У здоровых людей в плазме крови преобладает этантиол (80-90%) над метантиолом (10-20%), содержание которого повышается при печеночной недостаточности. Mеркаптаны подвергаются детоксикации в печени путем сульфатной конъюгации. Одним из продуктов конъюгации является диметилсульфид, который вместе с другими меркаптанами элиминируется легкими. Запах изо рта при печеночной недостаточности обусловлен наличием этих веществ в выдыхаемом воздухе. Изменение соотношения тиоловых компонентов объясняет некоторые отличия запаха у разных больных и в разное время. В условиях гипераммониемии снижается активность сульфатной конъюгации.
Синергический токсический эффект аммиака и меркаптанов у больных с нарушением функции печени и/или системным портальным шунтом может вызвать энцефалопатию и кому. Аммиак – ключевое промежуточное вещество азотистого обмена, основным источником которого является пищевой белок. Освобождение аммиака в кишечнике происходит с участием бактериальных аммиак-образующих ферментов, главным образом уреазы, расщепляющей мочевину до аммиака. Он всасывается и по системе воротной вены попадает в печень. При прохождении крови через печень из нее извлекается 70-80% аммиака, подвергающегося детоксикации с образованием мочевины. В небольших количествах аммиак образуется в процессе метаболизма в ткани мозга, почек, миокарда и других органах. Основной путь утилизации аммиака в клетках различных органов – синтез глутамина (мозг, почки, печень, мышцы, сердце), аминирование α-кетоглутарата. Гипераммониемия, не превышающая 25-50% по сравнению с верхней границей нормы, не приводит к развитию энцефалопатии. Лишь при превышении содержания в 1,5-2 раза наблюдается токсическое действие этого метаболита на ткань мозга.
Ткань головного мозга обладает высокой способностью образовывать глутамин, что является защитным механизмом, позволяющим избежать токсического действия значительных концентраций аммиака. При печеночной энцефалопатии содержание глутамина в спинномозговой жидкости и ткани мозга прогрессивно нарастает. Нейроны оказывается не в состоянии полноценно обезвреживать аммиак, и он проявляет свое токсическое действие непосредственно на клетки мозга. Механизм токсического действия аммиака на клетки, в частности на нейроны головного мозга, связан с нарушением энергетического обмена, так как в процессе детоксикации происходит интенсивное потребление α-кетоглутаровой кислоты и АТФ, то есть имеет место уменьшение синтеза и увеличение потребления АТФ. Дефицит энергетических фосфатов, необходимых для реполяризации нейронов и других функций ЦНС, приводит к глубоким неврологическим и энцефалопатическим расстройствам. Детоксикация значительных количеств аммиака сопровождается уменьшением уровня глутаминовой кислоты и α-кетоглутарата в тканях наряду с повышением глутамина. Снижение содержания глутаминовой кислоты, являющейся нейротрансмиттером, и увеличение глутамина, проявляющего слабый эффект центрального депрессанта, имеет значение в развитии печеночной энцефалопатии.
Накопление аммиака, изменение соотношения глутаминовой кислоты, α-кетоглутарата, глутамина в ткани мозга, в частности, в клетках дыхательного центра, способствует его активации. Это имеет важное клиническое значение, поскольку у большинства больных с печеночной недостаточностью обычно возникает гипервентиляция и респираторный алкалоз. Достаточно часто имеется сочетание респираторного и метаболического алкалоза. При нормальном значении ph существует равновесие между содержанием аммония (NH3, NH4+) в средах. Изменение pH, согласно «неионной диффузии аммиака»,стимулирует его миграцию из среды с более высоким pH. Поэтому при алкалозе концентрация аммиаки в клетках увеличивается, при ацидозе – снижается. У больных циррозом печени с выраженными явлениями портокавального шунтирования, портокавальным анастомозом экскреция аммиака с мочой повышается, а экскреция мочевины снижается.
Нарушения липидного обмена. В гепатоцитах синтез фосфолипидов угнетается, в связи с чем накапливаются нейтральные липиды (в норме их содержание не превышает 5% массы печени) и развивается жировая дистрофия печени, при которой содержание триглицеридов может достигать 80% массы печени.
Причины накопления триглицеридов:
- Нарушение белково-синтетической функции гепатоцитов;
- Угнетение формирования липопротеидных комплексов, поступающих в кровь;
- Повреждение систем биологического окисления и связанное с этим снижение катаболизма липидов в гепатоцитах.
В гепатоцитах ингибируется процессы эстерификации и синтеза холестерина, поэтому накапливается уксусная кислота, являющаяся субстратом для его образования. В больших количествах уксусная кислота проявляет цитотоксическое действие. Холестерин необходим для синтеза липидных структур мембран, желчных кислот. Роль желчных кислот в обмене холестерина значительна, поэтому различные нарушения метаболизма желчных кислот сопровождаются серьезными нарушениями обмена холестерина. В крови при печеночной недостаточности содержание эфира холестерина снижено, а уровень свободного холестерина повышен
В печени происходит детоксикация жирных кислот с короткой цепью (ЖККЦ), образующихся в кишечнике под влиянием бактериальной флоры. Они имеют в своем составе 4-8 углеродных атомов [бутановая (С4), валерьяновая (С5), капроновая (С6), каприловая (C8)]. У пациентов с печеночной комой уровень этих кислот возрастает в 3-8 раз по сравнению с нормой в связи с нарушением захвата и эстерификации ЖККЦ гепатоцитами. При печеночной недостаточности, не сопровождающейся комой, в крови определяется лишь незначительное их повышение. Утяжеление процесса характеризуется не только увеличением содержания ЖККЦ, но также повышением уровня жирных кислот с длинной цепью. Для головного мозга наиболее токсичны бутановая и изовалериановая кислоты. ЖККЦ транспортируются альбумином. При хронической печеночной недостаточности в условиях гипоальбуминемии жккц накаливаются в ткани мозга и синапсах. При избыточном образовании ЖККЦ связывающие способности альбумина могут быть исчерпаны. ЖККЦ ингибируют синтез мочевины и активность глутаминовой дегидрогеназы (два основных пути утилизации аммиака), нарастает гипераммониемия. Они обладают способностью потенцировать токсическое действие аммиака, и их синергический эффект оказывается значительно выше. ЖККЦ оказывают прямое воздействие на нейронные и синаптические мембраны, блокируя транспорт ионов на мембране нейрона и, соответственно, проведение импульса.
Генетический дефект метаболизма лейцина характеризуется повышением уровня изовалериановой кислоты (изовалериановая ацидемия). Прием белковой пищи определяет уровень этой жирной кислоты в крови. Клинически эта ферментопатия проявляется ступором. Описан генетический дефицит декарбоксилазы пропионат КоА. При этом в процессы метаболизма не включаются жирные кислоты с нечетным числом углеводных атомов (повышается уровень валериановой кислоты и других жирных кислот с нечетным числом атомов углерода). Развивается кетоновая гиперглицинемия, клинически проявляющаяся энцефалопатией. Для этих ферментопатий характерны гипераммониемия, повышение уровня жирных кислот с короткой цепью и развитие ступора.
При острой печеночной недостаточности, несмотря на обширный некроз гепатоцитов, содержание альбумина, холестерина, холинэстеразы и других веществ с большим периодом полураспада в плазме крови некоторое время существенно не меняется.
Нарушения углеводного обмена при патологии печени проявляется гипогликемией натощак вследствие истощения депо глюкозы в печени, нарушением липидного обмена, снижением способности организма поддерживать нормальный уровень глюкозы крови. Снижение содержания гликогена в гепатоцитах происходит в результате дисбаланса ферментативных систем: активации гликогенфосфорилазной системы и угнетения гликогенсинтетазы. Наиболее выраженное снижение уровня глюкозы определяется у больных с циррозом и синдромом рея. Для печеночной недостаточности характерно также нарушение процессов окисления глюкозы, глюконеогенеза, превращения галактозы и фруктозы в глюкозу. Изменение межуточного обмена при печеночно-клеточной недостаточности проявляется повышением содержания пировиноградной кислоты в сыворотке крови в 1,5-3 раза (норма 34,1-102,2 мкмоль/л). Это связано со снижением окисления ее в цикле трикарбоновых кислот и дефицитом кофермента А. Окисление пировиноградной кислоты идет по альтернативному пути, при этом образуются такие токсичные вещества, как ацетон и бутиленгликоль.
Date: 2015-09-18; view: 553; Нарушение авторских прав