Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Классическая гемофилия (гемофилия А)
|
|
Гемофилия А - наиболее распространенная наследственная коагулопатия, обусловленная дефицитом или молекулярной аномалией прокоагулянтной части фактора VIII (антигемофильного глобулика) с рецессивным, сцепленным с Х-хромосомой типом наследования.
Этиология и патогенез. Локализующийся в Х-хромосоме ген гемофилии передается от больного гемофилией всем его дочерям, и они в дальнейшем передают этот ген своим потомкам. Все сыновья бального остаются здоровыми, так как получают единственную Х-хромосому от здоровой матери. Женщины-кондукторы заболевания, имеющие вторую нормальную Х-хромосому, как правило, не страдают кровоточивостью, но активность фактора VIII у них снижена в среднем в 2 раза. Половина сыновей этих женщин, имеющих ген гемофилии, имеют шансы родиться больными (при равной возможности получить от матери патологическую или нормальную Х-хромосому), а половина дочерей - стать передатчицами болезни.
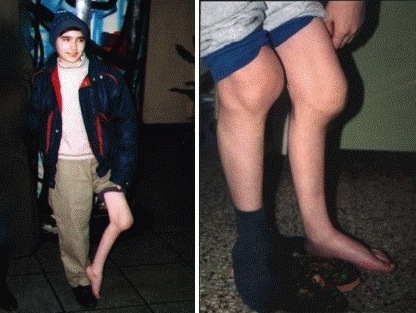
Клиническая картина. Геморрагический синдром при гемофилии отличается гематомным типом кровоточивости, для которого характерны обильные кровопотери, провоцируемые незначительными травмами. При рождении значительных геморрагических проявлений обычно не бывает. У взрослых больных возникают кровоизлияния в крупные суставы нижних и верхних конечностей, реже в мелкие суставы кистей и стоп, меж-позвоночные суставы. Острые гемартрозы рецидивируют, формируются хронические геморрагические деструктивные остеоартроэы.
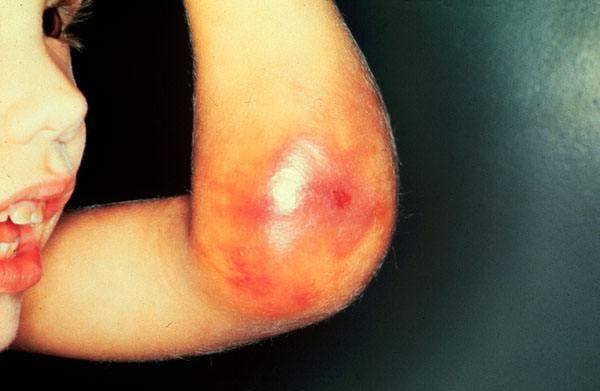
Часто отмечаются обширные подкожные, межмышечные, субфасциальные и забрюшинные гематомы, вызывающие некроз окружаюших тканей за счет сдавления питающих их сосудов. Инфицируясь, они могут быть причиной тяжелого сепсиса, при локализации в мягких тканях подчелюстной области, шеи, зева и глотки вызывают стеноз верхних дыхательных путей и асфиксию.
Серьезную опасность представляют рецидивирующие почечные кровотечения, сопровождаюшиеся приступами почечной колики.
Желудочно-кишечные кровотечения при гемофилии бывают спонтанными или обусловлены приемом лекарственных средств, вызывавших эрозию слизистых оболочек желудочно-кишечного тракта или обладающих антиагрегантными свойствами. Источником кровотечений могут быть язвы желудка и двенадцатиперстной кишки. Геморрагии в брыжейку, сальник, стенку кишки имитируют острые хирургические заболевания органов брюшной полости. Эффективность заместительной терапии служит диагностическим критерием в подобных случаях.
Для гемофилии характерны длительные кровотечения при травмах и операциях, поэтому- любое хирургическое вмешательство требует введения антигемофильных препаратов. Кровоизлияния в го.човной и спинной мозг и их оболочки почти всегда связаны либо с травмами, либо употреблением дезагрегантов.
Осложнения, встречающиеся при данном заболевании, подразделают на непосредственно связанные с геморрагиями (анемия, деструктивные процессы в костях, формирование гематом и псевдоопухолей, инфицирование их) и осложнения иммунного генеза (появление в крови больных в высоких титрах ингибиторои фактора VIII, а также РА, тромбоцитопения).
Диагноз и дифференциальный диагноз. Диагноз основывается на анамнестических данных, в частности наследственном анамнезе, наличии гематомного типа кровоточивости, результатах лабораторного исследования. Определение парциального тромбопластинового времени с кефалином подтверждает наличие гипокоагуляции, но имеет ориентировочное диагностическое значение. Тромбиновое и протромбиновое время не изменены. При тяжелых формах болезни отмечается увеличение общего времени свертывания плазмы, снижение потребления протромбина. Дефицит различных факторов свертывания и определение вида гемофилии проводят с помощью коррекционных проб. Выясняют, устраняется ли нарушение свертыванин компонентами крови с заведомо известным недостатком того или иного фактора. Если дефект свертывания, выявленный у больного, устраняется только адсорбированной сульфатом бария плазмой, в которой присутствует фактор VIII, но отсутствует фактор 1Х, то можно поставить диагноз гемофилии А. Если дефект исправляется только с помощью нормальной сыворотки, в которой присутствует фактор 1Х, но отсутствует фактор VIII, то ставят диагноз гемофилии В.
Вид гемофилии может быть определен также с помощью тестов смешивания. К плазме обследуемого добавляют последовательно в разных пробирках образцы плазмы больных с заведомо известной формой гемофилии, т. е. резким снижением уровней факторов VIII и 1Х, и определяют время свертывания (или время рекальцификации). При смешивании образцов с дефицитом одного фактора коррекции времени свертывания не происходит, в то время как смешивание образцов с дефицитом разных факторов свертывания ведет к взаимной коррекции этих факторов и нормализации процесса свертывания. Диагностика завершается количественным определением уровня дефицитного фактора.
Присутствие в плазме крови больного ингибитора фактора VIII:к доказывается с помощью гемагглютационного теста (проба Кумбса) с использованием антител против фактора VIII или теста, основанного на способности плазмы больного при добавлении к нормальной плазме удлинять парциальное тромбопластиновое время свертывания последней.
Дифференциальную диагностику следует проводить с болезнью Виллебранда и гемофилией В. В основе болезни Виллебранда - наследственного геморрагического диатеза с аутосомно-доминантным типом передачи лежит нарушение синтеза эндотелием сосуди-стой стенки крупномолекулярного компонента фактора VIII или ристоцетин-кофактора. Болеют лица обоего пола. Выраженность геморрагического синдрома при болезни Виллебранда разная - от сравнительно легких форм до тяжелых. Наиболее характерны носовые кровотечения, подкожные и внутрикожные геморрагии, но может наблюдаться и гематомный тип кровоточивости, как у больных гемофилией.
Диагноз при классическом варианте болезни Виллебранда устанавливают на основании следующих признаков: значительного увеличения времени кровотечения, снижения адгезии тромбоцитов к стеклу и коллагену, а также ристоцетин-агрегации, для которой также требуется VIII: ФВ, при нормальных других видах агрегации (под влиянием АДФ, адреналина, арахидоновой кислоты). Кроме того, отмечается снижение коагуляционной активности фактора VIII (уровня фактора VIII: к), устраняемого переливанием не только нормальной плазмы, но и плазмы больных гемофилией с постепенным, через 4 - 8 ч, нарастанием активности фактора VIII:к (из-за способности фактора Виллебранда, нормально присутствующего в переливаемой плазме, стимулировать синтез фактора VIII:к). Подтверждается диагноз определением уровня фактора Виллебранда в плазме больного по влиянию разных ее разведений на ристоцетин-агрегацию отмытых нормальных тромбоцитов и радиоиммунным методом.
Гемофилия В (болезнь Кристмаса) - наследственный геморрагический диатез, обусловленный дефицитом активности фактора 1Х (плазменного компонента тромбо-пластина). Передается по рецессивному, сцепленному с Х-хромосомой типу. Структурный ген фактора 1Х не связан с фактором VIII, так как располагается на другом конце Х-хромосомы. Ген фактора 1Х мутирует в 7 - 10 раз реже, чем ген фактора VIII, поэтому на долю болезни Кристмаса приходится 8 - 15 % всех случаев гемофилии. У большинства больных гемофилией В в плазме отсутствует антиген фактора 1Х, иммунные формы встречаются редко. Клиническая симптоматика, характер течения, возможные осложнения при болезни Кристмаса идентичны таковым при гемофилии А. В дифференциальной диагностике важно учитывать данные лабораторных исследований и тестов корреляции. Необходимо количественное определение фактора для оценки тяжести болезни.
Date: 2015-09-02; view: 1110; Нарушение авторских прав