Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Количественные нарушения аутосом
|
|
Количественные нарушения аутосом (как и половых хромосом) нормального кариотипа человека, являющиеся следствием ошибок, возникающих в процессе мейоза или митотического деления клеток, приводящих к неравномерному распределению хромосом материнской клетки между дочерними клетками, как правило, приводят к аномалиям развития.
Значительные нарушения развития, возникающие в результате увеличения количества хромосом диплоидного комплекса (2 n = 46) на число, кратное гаплоидному набору (п = 23), приводят к появлению полиплоидных особей, чаще всего триплоидов (3 n =69) либо тетраплоидов (4 n = 92), у человека обычно сопровождаются спонтанным выкидышем (абортом) на ранних этапах беременности.
Количественные изменения кариотипа человека чаще всего связаны с анеуплоидией (гетероплоидией), причём наибольшее распространение имеют трисомии, при которых в составе кариотипа появляется одна дополнительная хромосома, а также моносомии, связанные с потерей одной хромосомы. Возникающая при этом хромосомная патология, как правило, неизлечима, она имеет тяжелые клинические проявления, включая умственную отсталость.
К наиболее часто встречающимися хромосомным болезням человека, связанных с нарушениями числа аутосом, относится синдром Дауна (болезнь Дауна).Кариотип – 47, XX (XY) +21. Частота синдрома составляет 1:700 — 800 новорожденных.
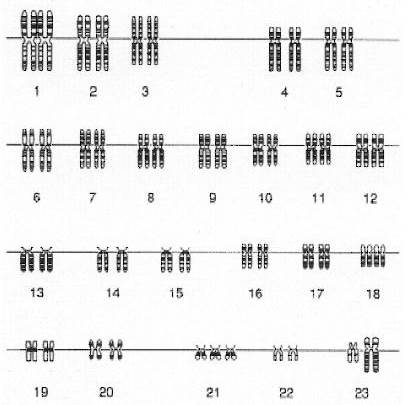
Дети с синдромом Дауна рождаются с умеренно выраженной пренатальной гипоплазией[2]. Течение беременности часто сопровождается токсикозом, угрозой выкидыша. Продолжительность беременности обычно не отличается от нормы.Внешними диагностическими признаками синдрома являются: широкое плоское лицо с широкой переносицей и монголоидным разрезом глазных щелей; характерны плоский затылок, приросшие мочки ушей, пальцы часто сращены, на ладонях одна поперечная линия (“обезьяннья”) вместо одной продольной и двух поперечных; рот полуоткрыт, конечности короткие. Для классической формы синдрома характерна выраженная умственная отсталость. Продолжительность жизни больных укороченная. Треть их умирает до конца первого года жизни, еще половина - до конца третьего года, а те, что выжили, стареют раньше, чем здоровые люди. Больные с классической болезнью Дауна бесплодные, поэтому они не передаются по наследственности.
Помимо простой трисомии по 21 паре хромосом, существуют транслокационная форма синдрома Дауна и мозаицизм, последний может быть представлен вполне нормальным фенотипом.Транслокационный вариант болезни, связанный с транслокацией 21 хромосомы на другую хромосому (у женщин - на 13-15, у мужчин - на 22) – две хромосомы фактически сливаются в одну, поэтому общее количество их не увеличивается, может передаваться по наследству.
Как правило, дети с синдромом Дауна чаще рождаются у женщин старше 35 лет (зависимость частоты рождения детей с синдромом Дауна от возраста матери показана на графике (рис.)).
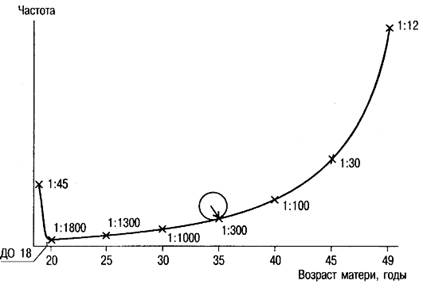
Рис. Зависимость частоты рождения детей с синдромом Дауна от возраста матери
Транслокационные формы, наоборот, чаще встречаются у молодых родителей. Мозаичные формы встречаются с одинаковой частотой во всех возрастных группах.
Второе место по частоте встречаемости после синдрома Дауна занимает синдром Патау –трисомия 13.Частота заболевания: 1 на 6000 рождений. Мальчики и девочки страдают этим заболеванием с одинаковой частотой. Имеет место простая полная трисомия 13 (47, XX (XY) + 13) как следствие нерасхождения этой пары хромосом в мейозе у одного из родителей; случаи, обусловленныетранслокациями; очень редко –мозаицизм и другие хромосомные варианты (изохромосома, инверсия).
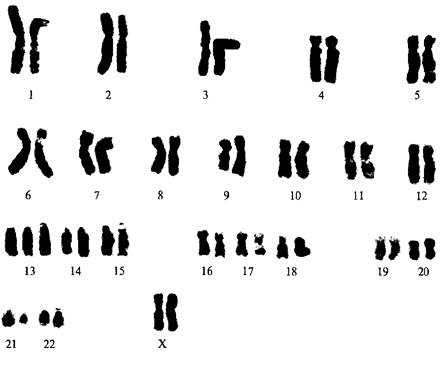
Рис. Кариотип при синдроме Патау
Типичными патологическими признаками синдромаПатауявляются истиннаяпренатальная гипоплазия (около 2500 г – средняя масса детей при рождении); аномалии черепа и лица – микроцефалия (иногда тригоноцефалия[3]), скошенный лоб, узкие глазные щели, гипотелоризм[4], запавшее переносье, низкорасположенные и деформированные ушные раковины;на коже головы имеются дефекты скальпа овальной или округлой формы, до 1 см в диаметре, дно таких дефектов представлено апоневротическим шлемом; характерны также расщелина губы и нёба и полидактилия. В сочетании с указанными выше признаками у определённого процента больных отмечаются врожденные пороки, пищеварительного тракта, почек,половых органов, органов зрения (анофтальмия, микрофтальмия, дисплазии сетчатки, колобома радужки, помутнение хрусталика). Центральная нервная система поражается в 100% случаев. Все дети с синдромом Патау имеют тяжелую умственную отсталость (глубокая идиотия). На первом году жизни умирают 95% больных с синдромом Патау (более половины из них – в перинатальном периоде).
Синдром Эдвардса –трисомия по 18-й хромосоме (рис. Х.7). Кариотип: 47, XX (XY) + 18. Как и при синдроме Дауна, выявляется связь между частотой трисомии с возрастом матери и дополнительная хромосома, как правило, имеет материнское происхождение.
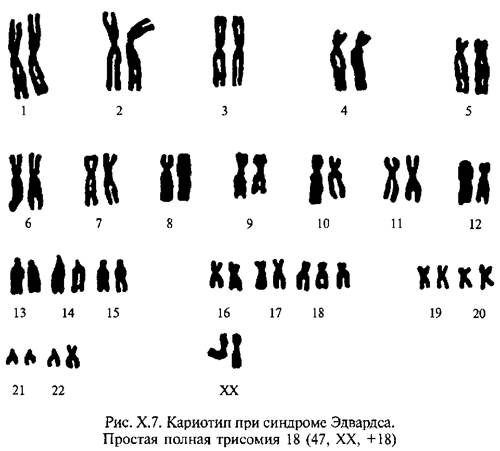
Частота синдрома 1:7000; девочки болеют примерно в три раза чаще мальчиков. Среди специфических клинических проявлений: долихоцефалия, гипоплазия нижней челюсти и микростомия, узкие и короткие глазные щели, маленькие низко расположенные ушные раковины, характерное сгибательное положение пальцев кисти, аномально развитая стопа (пятка выступает и свободно провисает, первый палец короче второго), выступающий затылок; практически постоянны пороки сердца и крупных сосудов (основная причина высокой летальности на первом году жизни), часты пороки желудочно-кишечного тракта, пороки почек и половых органов; в ряде случаев, - спинно-мозговая грыжа и расщелина губы.Все выжившие больные имеют глубокую степень умственной отсталости (идиотию).
Диагностика синдрома Эдвардса особенно затруднена, однако косвенными признаками по данным УЗИ, указывающими на синдром Эдвардса у плода, могут стать малая величина плаценты, недоразвитие или отсутствие одной из пупочных артерий в пупочном канатике.
Помимо рассмотренных примеров известны также трисомии хромосом 19 и 20, которые в 50 % случаев приводят к летальному исходу в возрасте до двух месяцев. Трисомии по другим хромосомам можно обнаружить примерно в 2,8 % случаев такой патологии и все они, как правило, заканчиваются гибелью плода до рождения. Увеличение числа хромосом второй и третьей пар несовместимо с формированием эмбриона.
При синдроме Варкани наблюдается трисомия 8. Кариотип: 47, XX (XY) + 8. Частота – не более, чем 1:5000. Среди больных преобладают мальчики (соотношение мальчиков и девочек 5:2). Клиническая картина синдрома описана у детей с отставаниями в умственном развитии, отклонениями в строении лица (выступающий лоб, косоглазие, эпикант, глубоко посаженные глаза, гипертелоризм глаз, толстые губы, вывернутая нижняя губа, большие ушные раковины с толстой мочкой), пороками в строении опорно-двигательного аппарата (как добавочные позвонки, неполное закрытие позвоночного канала, аномалии формы и положения рёбер или добавочные рёбра, отсутствие надколенников иконтрактуры суставов), мочевой системы и другими врождёнными пороками развития. Полные трисомии 8, как правило, летальны. Большинство описанных случаев (около 90%) относится к мозаичным формам. Трисомия 8 — результат вновь возникшей мутации (нерасхождение хромосом) на ранних стадиях бластулы, за исключением редких случаев новой мутации в гаметогенезе. Прогноз физического, психического развития и жизни неблагоприятный, хотя описаны пациенты в возрасте 17 лет.
Date: 2015-09-02; view: 1252; Нарушение авторских прав