Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Наследственные нарушения обмена липидов
|
|
Из наследственных (врождённых) нарушений метаболизма липидов в этом разделе будут рассмотрены нарушения обмена холестерола – (семейная гиперхолестеролемия) и группа наследственных заболеваний, связанных с нарушением обмена сложных липидов и накоплением их в различных органах и тканях (лизосомные болезни накопления).
Нарушения обмена холестерола.
В данном разделе будет рассмотрена патология обмена холестерола относящаяся к первичным (наследственным) дислипопротеинемиям. Наиболее тяжёлой формой наследственной дислипопротеинемии является семейная гиперхолестеролемия, обусловленная увеличением содержания общего холестерола в крови (для гомозигот до 20,3 ммоль/л, для гетерозигот до 10,8 ммоль/л).
Семейная гиперхолестеролемия (недостаточность рецепторов для липопротеинов низкой плотности - ЛПНП, наследственная гиперхолестеролемия) наследуется по аутосомно-доминантный типу. Молекулярный дефект, определяющий развитие этого заболевания, был описан Брауном (Brown M. S., 1981) и оказался связанным с дефектом рецепторного белка, участвующего в поглощении ЛПНП из крови.**
**Локализуется рецептор на поверхности гепатоцитов и связывает липопртотеины низкой плотности (ЛПНП), холестерол-транспортные белки, обеспечивая их проникновение внутрь клетки путем эндоцитоза. В клетке ЛПНП распадаются внутри лизосом и высвобождают холестерол, который может быть использован для синтеза стероидных гормонов, липидов мембран. Мутации гена, кодирующего рецептор ЛПНП, локализованного в коротком плече 19 хромосомы, приводят к нарушению процессов деградации ЛПНП. Первая мутация в гене рецептора ЛПНП была выявлена Wingerde в 1981 году.
Поскольку поступление холестерола ЛПНП в клетки снижено, его содержание в плазме крови быстро нарастает. Повышенная концентрация холестерола может быть обнаружена ещё у детей в пуповинной крови. У гомозигот рецепторы для ЛПНП фактически отсутствуют и концентрация холестерола ЛПНП в плазме крови превышает величины, характерные для здоровых лиц более чем в 3 раза. Концентрация триглицеридов остается в пределах нормы.
Из наследственных нарушений метаболизма это патологическое состояние характеризуется особенно высокой смертностью. В связи с развитием сердечно-сосудистых заболеваний возраст таких пациентов редко превышает 20 лет. У гетерозиготных носителей аномального гена число рецепторов для ЛПНП снижено приблизительно на 50% и содержание холестерола ЛПНП в плазме крови приблизительно в 2 раза выше, чем у здоровых людей. У этих больных степень риска развития сердечно-сосудистых заболеваний повышается в 10 раз. У гомозигот ксантомы * сухожилий и ксантелазмы* развиваются в раннем детстве, но у гетерозиготных носителей — только после 20 лет.
*Ксантома (xanthoma) - возникает в коже при нарушении холестеринового обмена. Множественная узелковая ксантома наблюдается чаще на разгибательных участках конечностей, волосистой части головы, узелки, размером от макового зерна и больше, отличаются своим золотисто-желтоватым цветом и розовато-фиолетовым венчиком по периферии, имеют мягкую, плотную или келоидную консистенцию. Течение хроническое. Ксантелазма (xanthelasma) - это плоская ксантома, наблюдается чаще у женщин, страдающих диабетом, гиперхолестеролемией, как правило, располагается на веках в виде небольших желтоватых бляшек, несколько возвышающихся над кожей. Могут располагаться группами, спонтанного исчезновения не происходит.
В семьях с моногенным типом наследования имеется четкое различие между клинически здоровыми гомозиготными и гетерозиготными индивидуумами, в противоположность тому, что имеет место при более распространенном полигенном типе заболевания. В большинстве стран моногенная гиперхолестеролемия составляет менее 5% всех случаев первичной гиперхолестеролемии.
Наследственные заболевания, связанные с отложением липидов в различных органах и тканях – это группа болезней, в основе которых лежит накопление избыточных количеств фосфолипидов и сфинголипидов* в клетках паренхиматозных органов и нервной ткани.
*Сфинголипиды являются структурными компонентами клеточных мембран, в частности, миелиновых оболочек нервных волокон, поэтому нарушение постоянно протекающих процессов синтеза, распада этих липидов в лизосомах клеток создает патологическую основу поражения большинства жизненно важных органов, включая серое и белое вещества головного мозга. Дефекты распада сфинголипидов связаны с недостаточностью соответствующих ферментов.
К ним относят большую группу наследственных заболеваний получивших название сфинголипидозы (табл. 1). В клиническом аспекте сфинголипидозы характеризуются прогрессирующими умственными и двигательными расстройствами, поражением костной системы, паренхиматозных органов (печень, селезёнка, почки), кожи и сетчатой оболочки глаза.
Табл.1
Сфинголипидозы человека.
| Название заболевания | Недостаточность фермента | Накапливающийся сложный липид |
| Фукозидоз | альфа-Фукозидаза | Cer-Glc-GalNAc-Cal-:-Fuc Н-Изоантиген |
| Генерализованный ганглиозидоз | GM1-бета-Галактозидаза | Cer-Glc-Gal(NeuAc)-GalNAc-:-Gal Ганглиозид GM1 |
| Болезнь Тея-Сакса | Гексозаминидаза А | Cer-Glc-Gal-(NeuAc)-:-GalNAc Ганглиозид GM2 |
| Вариант болезни Тея-Сакса, или болезнь Сандхоффа | Гексозаминидазы А и В | Cer-Glc-Gal-Gal-:-GalNAc Глобозид + ганглиозид GM2 |
| Болезнь Фабри | альфа-Галактозидаза | Cer-Glc-Gal-:-Gal Глоботриаозилцерамид |
| Церамидлактозидлипидоз | Церамидлактозидаза (бета-галактозидаза) | Cer-Glc-:-Gal Церамидлактозид |
| Метахроматическая лейкодистрофия | Арилсульфатаза | Cer-Gal-:-OSO3 3-Сульфогалактозилцерамид |
| Болезнь Краббе | бета-Галактозидаза | Cer-:-Gal Галактозилцерамид |
| Болезнь Гоше | бета-Глюкозидаза | Cer-:-Glc Глюкозилцерамид |
| Болезнь Нимана-Пика | Сфингомиелиназа | Cer-:-P-холин Сфингомиелин |
| Болезнь Фарбера | Церамидаза | Ацил-:-Сфингозин Церамид |
Сфинголипидозы - врожденные нарушения метаболизма сфинголипидов, обусловленные отсутствием лизосомных ферментов, катализирующих процессы их распада, среди этой группы будут рассмотрены, следующие заболевания:
1. Болезнь Гоше;
2. Болезнь Нимана-Пика;
3. Болезнь Тея-Сакса.
Болезнь Гоше. Впервые заболевание описал в 1882 году французский студент медик Филипп Шарль Эрнст Гоше (Gaucher P. C. E.). Внутриклеточный липоидоз, характеризующийся накоплением цереброзидов в клетках ретикулоэндотелиальной системы – макрофагах, которые принимают соответствующую морфологию, получивших название клеток Гоше или «клеток накопления», (рис. 9). Болезнь Гоше встречается с частотой от 1:40000 до 1:60000 у представителей почти всех этнических групп, а в популяции евреев Ашкенази частота этого заболевания достигает 1:450.
Накопление цереброзидов (глюкоцереброзида) в клетках обусловлено наследственным дефицитом глюкоцереброзидазы * (glucocerebrosidase, КФ 3.2.1.45) - лизосомного фермента катализирующего гидролитический распад β-D-глюкозилцерамида на β-D-глюкозу и церамид. Заболевание наследуется по аутосомно-рецессивному типу.
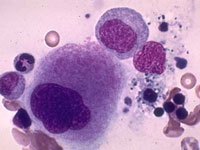
Рис. 9. Клетки Гоше.
*Ген глюкоцереброзидазы локализуется на 1 хромосоме человека в позиции q21 (Cormand B.,1997). Недостаточность энзима обусловлена различными мутациями гена. Первая мутация в гене глюкоцереброзидазы человека была выявлена в 1987 году Tsuji S. Список идентифицированных мутаций в гене глюкоцереброзидазы представлен missense мутациями, nonsense мутациями, сплайсинговыми мутациями, а также делециями и инсерциями. Интенсивный поиск и открытие мутаций данного гена было осуществлёно японскими, американскими исследователями за период с 1989 по 1997 гг. Основные проявления болезни Гоше обусловлены накоплением клеток, перегруженных «шлаками» и нарушением функции этих клеток. Накопление этих клеток в различных органах приводит к увеличению их размеров (увеличение печени и селезёнки) и нарушению структуры и функции этих органов. Критическим при этом заболевании является нарушение функции макрофагов, перегруженных «шлаками», что приводит к анемии, кровоточивости (тромбоцитопения), частым переломам костей из-за их хрупкости. Это связано с тем, что макрофаги в организме регулируют многие жизненно важные процессы: кроветворение, свертывание крови, обмен костной ткани и многое другое. Основные симптомы у больных с болезнью Гоше - это увеличение печени и селезёнки, анемия, тромбоцитопения, хронические боли в костях (остеоалгии), повышение температуры – лихорадка, поражение суставов и частые переломы.Клинически различают три типа болезни Гоше: тип I (взрослый тип), тип II (инфантильный тип), тип III (ювенальный тип).
Диагноз болезни Гоше устанавливается на основании биохимического аналаза включающего определение активности глюкоцереброзидазы в лейкоцитах крови. Снижение активности кислой глюкоцереброзидазы менее 30% от нормального уровня является основанием для биохимического подтверждения диагноза, основанного на клинических данных. Характерным биохимическим маркёром болезни Гоше является повышение активности хитотриозидазы в сыворотке крови больных в 1000 и более раз.
Болезнь Нимана-Пика. Болезнь Ниманна — Пика (англ. Niemann-Pick disease) — это наследственное заболевание, в основе которого лежит недостаточность активности кислой сфингомиелиназы* (сфингомиелинфосфодиестераза, acid sphingomyelinase, КФ 3.1.4.12). Данный лизосомный фермент катализирует реакцию распада сфингомиелина на церамид и фосфохолин в мембранах лизосом. Его недостаточность приводит к избыточному накоплению сфингомиелина, а как следствие к нарушению липидного метаболизма, включая накопление холестерола и других клеточных липидов. Заболевание наследуется по аутосомно-рецессивному типу. Различают три типа данного заболевания: типы A, B связаны с недостаточной активностью кислой сфингомиелиназы, а тип C обусловлен мутациями генов NPC1 или NPC2, которые кодируют белок клеточной мембраны, отвечающей за транспорт холестерола и липидов внутри клетки.
* Мутации гена кислой сфингомиелиназы приводят к недостаточной активности данного энзима. Ген локализуется на 11 хромосоме человека в позиции p15.4-p15.1. В 1992 году Schuchman клонировал и секвенировал ген сфингомиелиназы. Кодирующая область гена состоит из 1116 нуклеотидов, некодирующие 5' и 3'области состоят из 468 нуклеотидов. Ген разделяется 5 интронами на 6 экзонов. Размер экзонов варьирует от 77 до 773 пар нуклеотидов, а интронов от 153 до 1059 пар нуклеотидов. Регуляторная область гена включает отдельные промоторные элементы - четыре SP-1 связывающих сайта, два TATA-box, два CAAT-box, два NF-1 и один AP-1 связывающих сайта. Во втором интроне локализуется Alu элемент. Интригующим обстоятельством в строении данного гена является наличие трех дополнительных открытых рамок считывания (ORFs), предположительно кодирующих полипептиды из 101, 104, и 158 аминокислот
Тип А — самый тяжёлый тип, который дебютирует у детей в грудном возрасте и характеризуется увеличением печени и селезёнки (гепатоспленомегалия), прогрессирующим поражением нервной системы. Характеризуется высокой смертностью детей в младенчестве. Наиболее часто встречается этот тип болезни Ниманна-Пика у выходцев из центральной и восточной Европы (Ашкенази) — примерно 1:40000.
Тип B - умеренный, проявляется гепатоспленомегалией, задержкой роста и нарушением лёгочной функцией с развитием респираторных инфекций. Другие показатели включают повышенный уровень холестерола и липидов в крови, тромбоцитопению. Больные чаще доживают до взрослого возраста.
Тип С - проявляется в грудном возрасте или у взрослых. Симптомы включают тяжёлые печёночные нарушения, проблемы с дыханием, задержку в развитии, припадки, повышенный мышечный тонус (дистония), нарушение координации движения, питания и движения глаз в вертикальной плоскости. Больные доживают до взрослого возраста. Частота заболевания — 1:150000.
Болезнь Тея-Сакса. Болезнь Тея-Сакса или GM2-ганглиозидоз (Tay-Sachs disease) - наследственное заболевание с преимущественным поражением нервной системы, характеризуется прогрессирующим снижением зрения в сочетании с деградацией интеллекта до идиотии и разнообразными неврологическими расстройствами. Первые случаи этого заболевания описаны Tay в 1881 году и Sachs в 1887 году. Заболевание проявляется в виде двух форм: тип 1 (инфантильный), который заканчивается летальным исходом ещё в младенчестве, тип 2 (ювенальный), характеризуется более благоприятным прогнозом и большей выживаемостью больных до зрелого возраста. Заболевание наследуется по аутосомно-рецессивному типу.
Болезнь Тея-Сакса обусловлена отсутствием активности гексозаминидазы * (beta-hexosaminidase, beta-N-acetyl-hexosaminidase, КФ 3.2.1.52) в клеточных лизосомах. Данный энзим катализирует катаболизм GM2-ганглиозида. У больных при этом заболевании происходит накопление субстрата реакции - GM2-ганглиозида в основном клетках центральной нервной системы.
*Отсутствие активности данного энзима обусловлено мутациями гена гексозаминидазы локализованного на 15 хромосоме человека в позиции q23-q24. В настоящее время за 10 летний период изучения мутационного поражения в гене гексозаминидазы человека было выявлено более 40 мутаций (идентифицированы missense, nonsense мутации, инсерции).
 Приложение
Приложение
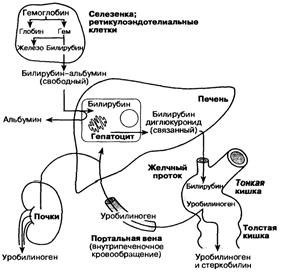
Схема 1. Обмен билирубина и его экскреция печенью. Свободный билирубин (непрямой), связанный билирубин (прямой).
.
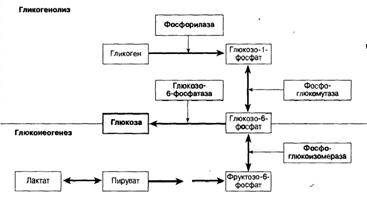
Схема 2. Образование глюкозы при гликогенолизе и глюконеогенезе. Глюкозо-6-фосфат является важным промежуточным метаболитом образования глюкозы как при гикогенолизе, так и при глюконеогенезе.
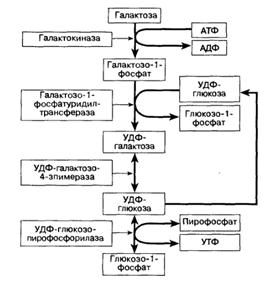
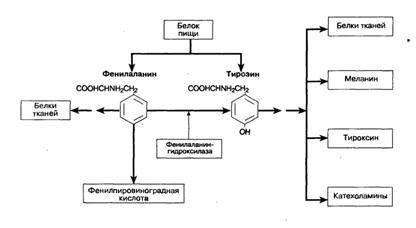 Схема 3. Метаболический путь превращения галактозы в глюкозу. УДФ- уридиндифосфат, УТФ- уридинтрифосфат.
Схема 3. Метаболический путь превращения галактозы в глюкозу. УДФ- уридиндифосфат, УТФ- уридинтрифосфат.
.
Схема 4. Метаболический путь превращения фенилаланина в тирозин. Показан участок действия фенилаланингидроксилазы, фермента дефицит которого наблюдается при фенилкетонурии.
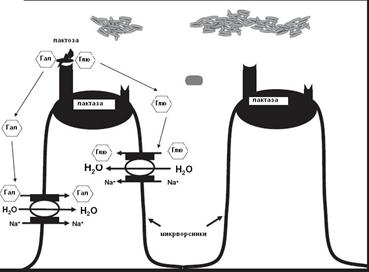
Схема 5. Расщепление дисахарида лактозы ферментом из класса гидролаз - лактазой (КФ3.2.1.108) локализованного в клеточной мембране энтероцитов в норме. (Глю) - глюкоза, (Гал)- галактоза.
Список литературы
Биохимия. Учебник для ВУЗов. /Под ред. Е. С. Северина. – М.: ГЭОТАР-МЕД, 2003. – 784с.
Березов Т.Т., Коровкин Б.Ф. Биологическая химия. Изд. 3-е. – М.: Медицина, 1998. – 704 с.
Интернет ресурсы: www. medbiol.ru, www.brenda-enzymes.info/index.php4.
Элиот В., Элиот Д. Биохимия и молекулярная биология. – М.: НИИ биомед. Химии РАМН, 2000. – 372 c.
Boll W., Wagner P., Mantei N. Structure of the chromosomal gene and cDNAs coding for lactase-phlorizin hydrolase in humans with adult-type hypolactasia or persistence of lactase. // Am. J. Hum. Genet. 1991., V.48., P. 889–902.
Gremse D.A., Bucuvalas J. C,Baluster W. F. Efficacy of cornstarch therapy in type III glycogen-storage disease. // Am. J. Clin.Nutr. 1990., V.52 (4)., P.671-674
Lomer M. C. E., Parkes G. C., Sanderson J. D. Review article: lactose intolerance in clinical practice – myths and realities. // Aliment Pharmacol Ther., 2008., V. 27, P. 93–103.
Mistry P. K., Cox T. M. The glucocerebrosidase locus in Gaucher's disease: molecular analysis of a lysosomal enzyme.// J. Med. Genet., 1993., V. 30., P. 889-894.
Naim H. Y. Molecular and cellular aspects and regulation of intestinal lactase-phlorizin hydrolase. // Histol Histopathol. 2001., V.16., P. 553–561.
Schuchman E. H. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann–Pick disease.// J. Inherit. Metab. Dis., 2007., V. 30., P. 654–663.
Tropak M. B., Reid S. P., Guiral M., Withers S. G., Mahuran D. Pharmacological enhancement of β-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff patients. // The Journal of Biological Chemistry., 2004., V. 279, N.14, Issue 2, P.13478–13487.
Date: 2015-05-23; view: 2418; Нарушение авторских прав