Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Кислота ацетилсалициловая
|
|
6. Получают взаимодействием салициловой кислоты с уксусным ангидридом и взаимодействием фенолята натрия с углекислым газом.
7.

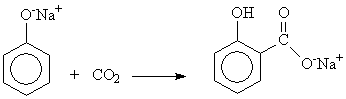
8. Схема получения аспирина в промышленности:

9. Кислота ацетилсалициловая является веществом органического происхождения.
10. Мало растворима в воде, легко растворим в 96 % спирте.
ЭТАП
1. 4-гидроксибензойная кислота, 4-гидрокисизофталевая кислота, ацетилсалициловая кислота
2. Жидкостная хроматография с УФ-детектором по ГФ РК 2.2.29.
3. Не более 2·10-3% (20 млн.-1). 1.0 г субстанции растворяют в 12 мл ацетона Р и доводят объем раствора водой Р до 20 мл. 12 мл полученного раствора должны выдерживать испытание на тяжелые металлы. ГФ РК Т II, частная статья «Ацетилсалициловая кислота» (с.121).
4. Не более 0,1 %. Определение проводят из 1.0 г субстанции. ГФ РК Т II, частная статья «Ацетилсалициловая кислота» (с.121).
5. Не более 0,5 %. Определение проводят из 1.0 г субстанции. ГФ РК Т II, частная статья «Ацетилсалициловая кислота» (с.121).
ЭТАП
5. Идентификация проводится по двум методам: первая идентификация: А, В (физико-химические методы, качественная реакция) и вторая идентификация: В, C, D (физико-химический метод и качественные реакции).
6. Первая идентификация: А, В
А. Инфракрасный спектр поглощения (абсорбционная спектрофотометрия в инфракрасной области, 2.2.24 ГФ РК, Т 1) субстанции должен соответствовать стандартному спектру СО ГФ РК кислоты ацетилсалициловой.
В. Химическая реакция с раствором натрия гидроксида разбавленного, кислотой серной разбавленной. Выпадает кристаллический осадок.
Температура плавления полученного остатка (2.2.14) должна быть от 156 до 161 °С.
7. Вторая идентификация: В, C, D
В. См пункт 2
С. Химическая реакция с кальцием гидроксида Р, раствором нитробензальдегида Р, кислотой хлороводородной разбавленной Р. Бумага окрашивается в голубой цвет.
D. Химическая реакция на салицилаты.
8. Идентификация по методике С. кальция гидроксид Р, раствор нитробензальдегида Р, кислота хлороводородная разбавленная Р.
В пробирке смешивают 0,1 г субстанции с 0,5 г кальция гидроксида Р. Смесь нагревают и в появляющихся парах выдерживают кусочек фильтровальной бумаги, пропитанный 0.05 мл раствора нитробензальдегида Р. Постепенно бумага окрашивается в желтовато-зеленый или голубовато-зеленый цвет. Затем бумагу смачивают кислотой хлороводородной разбавленной Р, бумага окрашивается в голубой цвет.
ЭТАП
6. Метод титрования.
1.000 г субстанции помещают в колбу с притертой стеклянной пробкой, растворяют в 10 мл 96 % спирта Р и прибавляют 50.0 мл 0,5 М раствора натрия гидроксида. Колбу закрывают и выдерживают в течение 1 ч. Полученный раствор титруют 0.5 М кислотой хлороводородной, используя в качестве индикатора 0,2 мл раствора фенолфталеина Р.
7. Титрант: 0,5 М кислота хлороводородная.
Индикатор: Фенолфталеин.
Изменение окраски индикатора при рН 3,1-4,0.
Розовое окрашивание.
8. Препарат растворяют в 10 мл 96% спирта Р. Спирт должен быть охлажден до 8-10°С (для предотвращения гидролиза препарата при титровании титрантом).
9. Расчет содержания лекарственного вещества в субстанции, в процентах (Х):
V·T·Kn·100
Х (%) = a,
где V – количество тированного раствора, израсходованного на титрование лекарственного вещества;
Т – титр, количество растворенного вещества, соответствующего 1 мл титрованного раствора, г.;
а – навеска порошка, взятая для анализа, г;
Kn– коэффициент поправки.
10. 1 мл 0,5 М раствора натрия гидроксида соответствует 45,04 мг С9Н8О4
ЭТАП
5. Лекарственное вещество выпускается в форме таблеток, таблеток шипучих, таблеток, кишечнорастворимых для приема внутрь.
6. По 10 таблеток в контурную ячейковую упаковку из пленки поливинилхлоридной по ГОСТ 25250-88 и фольги алюминиевой по ГОСТ 745-2003.
По N контурных упаковок вместе с инструкцией по медицинскому применению на государственном и русском языках помещают в коробку из картона по ГОСТ 7933-89.
Маркировка упаковки на государственном и русском языках в соответствии с Техническим регламентом «Требования к безопасности лекарственных средств», утверждено постановлением ПравительстваРеспублики Казахстанот 14 июля 2010 года № 712.
7. Препарат применяется в качестве противоревматического, противовоспалительного, болеутоляющего, жаропонижающего средства.
8. Хранят в хорошо укупоренной таре в защищенном от света месте.
МАГНИЯ СУЛЬФАТ
6. Магния сульфат получают нагреванием магнезита с избытком серной кислоты (избыток кислоты необходим чтобы избежать образования остатков солей магния).
7. Химические реакции получения магния сульфата:
MgCO3 + H2SO4 = Mg SO4 + H2O + СO2
Mg SO4 * 7 H2O = Mg SO4 + 7 H2O
MgCO3 +(NH4)2SO4 = Mg SO4 + NH3 + H2O+ СO2
8. Химическая схема производства сульфата магния в промышленности:

9. Магния сульфат - вещество неорганической природы, относится к классу неорганических солей.
10. Представляет собой бесцветные призматические кристаллы, выветривающиеся на воздухе.
ЭТАП
7. Указал образование белого кристаллического осадка двойного фосфата аммония и магния при взаимодействии препарата с аммония фосфатом натрия в аммиачном растворе в присутствии хлорида аммония. Осадок нерастворим в разведенных минеральных кислотах и уксусной кислоте.
Определил ион магния с 8-оксихинолином в присутствии аммиачного раствора с добавлением хлорида аммония. Магния оксихинолят окрашен в зеленовато – желтый цвет.
8. Сульфат – ион открыл раствором хлорида бария: выпадает белый осадок сульфата бария, нерастворимого в минеральных кислотах.
9. Объяснил, что во избежание образования в щелочной среде аморфного осадка гидроксида магния используют раствор хлорид аммония. Недостаток: избыток хлорида аммония может препятствовать осаждению фосфата магния – аммония.
10. Подобрал растворы: фосфат натрия, хлорид аммония, аммиака, кислоты хлороводородной, бария хлорида, 8 – оксихинолин, кислота уксусная.
11. Выбрал пипетки на 1, 2 мл, пробирки, весы ручные, разновесы, бюкс. Выбрал растворы хлорида аммония, фосфата натрия, аммиака, хлороводородной кислоты, бария хлорида.
12. К 2 мл раствора магния сульфата (0.005 – 0.05 г иона сульфата) прибавлял 0.5 мл разведенной хлороводородной кислоты и 0.5 мл раствора бария хлорида: образуется белый осадок, нерастворимый в разведенных кислотах.
ЭТАП
7. Растворил 2 г препарата в 20 мл воды очищенной. После 5 минутного кипячения раствора сравнивают с водой очищенной на компоратаре. Прозрачным считают раствор, если при рассмотрении невооруженным глазом не наблюдается присутствие нерастворимых частиц. Прибавил к 5 мл раствора препарата (2:20) 5 мл воды очищенной и 2 капли раствора фенолфталеина; раствор должен быть бесцветным. Розовое окрашивание появилась от добавления 0.1 мл 0.01 н раствора натрия гидроксида. Пробирки одинакового диметра, стекла; пипетки на 1.2, 5, 10 мл, эталонный раствор Б на хлориды. Вода очищенная, раствор кислоты азотной, серебра нитрата, 0.1 н раствор натрия нитрита, 20 % раствор персульфата аммония, 0.01 н калия перманганата.
8. Назвал метод количественного определения магния сульфата – комплексонометрия. Выбрал титрант 0.05 М раствор трилона Б. Выбрал индикатор кислотный хром черный специальный.
9. Указал изменение окраски в эквивалентной точке – от красной к синей. Содержание препарата должно быть не менее 99.0 % и не более 102.0 %.
10. Добавил аммиачный-буферный раствор. Указал рН 9.5-10 титруемого раствора.
11. Применил индикатор в смеси с натрия хлоридом (1:100). Указал образование комплексов металлов с эриохром черным специальным в щелочном растворе при рН 7.0-11.0.
12. Указал на малую устойчивость раствора индикатора во времени.
ЭТАП
6. Э=М.М.
7. Т=246*48*0.05 / 1000= 0.01232
8. Расчет содержания лекарственного вещества в препарате, в процентах (Х):
Х(%)= V*Т*100* Kn / а
где V - количество титрованного раствора, израсходованного на титрование лекарственного вещества, мл;
Т- титр, количество растворенного вещества, соответствующего 1 мл титрованного раствора, г/мл.
а – навеска порошка, взятая для анализа, г.
Kn - коэффициент поправки.
9. Расчет содержания лекарственного вещества в растворе для инъекций, (Х) в пересчете на один мл.
Х(г)= V*N*Kn*1 / а;
где а - объем раствора исследуемого раствора, мл;
V - количество титрованного раствора, израсходованного на титрование лекарственного вещества, мл;
Т- титр, количество растворенного вещества, соответствующего 1 мл титрованного раствора, г/мл.
Kn - коэффициент поправки.
10. Содержание магния сульфата в 25 % растворе для инъекций должно быть в пределах 0.242-0.258 г в 1 мл.
ЭТАП
5. Указал, что 10 ампул помещают в коробки из картона коробочного марки А по ГОСТ 7933-75. Указал, что в коробки вкладывают скарификатор ампульный по ТУ 64-1-994-80 или нож ампульный по ТУ 400-6-169-85.
6. Объяснил, что на каждой ампуле методом глубокой печати быстро закрепляющейся краской по ТУ 788-86 указывают название препарата на государственном и русском языках, концентрацию раствора в %, объем в мл, номер серии и срок годности.
7. Транспортирование проводят в соответствии с ОСТ 64-034-87 и ГОСТ 17768-80.
8. В защищенном от света месте, срок годности растворов по 5 и 10 мл, 2 года.
НОВОКАИН
1. Новокаин получают переэтерификацией бензокааина β-диэтиламиноэтанолом.
2. Химическая реакция получения новокаина:

3. Технологическая схема получения новокаина:

4. Вещество органической природы. Аминокислота ароматического ряда. Лекарственный препарат, производное п-аминобензойной кислоты.
5. Температура плавления новокаина 154-158 оС.
ЭТАП
1. Прозрачность. Препарат должен быть прозрачным по сравнению с водой Р (ГФ РК I, т. 1, 2.2.1).
2. Цветность. Препарат должен быть бесцветным по сравнению с водой Р (ГФ РК I, т. 1, 2.2.2).
3. рН. От 3.8 до 4.5 (потенциометрически; ГФ РК I, т. 1, 2.2.3).
4. Механические включения. Испытания проводят методом ГФ РК I, т. 1, 2.9.20.
Препарат должен выдерживать требования Инструкции МЗ РК "По контролю инъекционных растворов на механические включения" И взамен И 42-3-85, г. Алматы, 1997 г.
В одной ампуле допускается наличие частиц размером более 10 мкм не более 6000 и частиц размером более 25 мкм – не более 600 (ГФ РК I, т.1, 2.9.19, метод I).
5. Родственные примеси. Испытания проводят методом тонкослойной хроматографии (ГФ РК I, т. 1, 2.2.27).
Препарат – испытуемый раствор.
На линию старта хроматографической пластинки cо слоем силикагеля GF254 P наносят 20 мкл (100 мкг) испытуемого раствора, 5 мкл (100 мкг) раствора сравнения (а), 5 мкл (0,5 мкг) раствора сравнения (b), 5 мкл (0,5 мкг) раствора сравнения (с) и в одну точку: 5 мкл (100 мкг) раствора сравнения (а), 5 мкл (0,5 мкг) раствора сравнения (b) и 5 мкл (0,5 мкг) раствора сравнения (с).
Пластинку с нанесенными пробами помещают в камеру с системой растворителей бензол Р – ацетон P (8: 2) и хроматографируют восходящим способом. Когда фронт растворителей пройдет 15 см от линии старта, пластинку вынимают из камеры, сушат на воздухе в течение 10 мин и просматривают в УФ-свете при длине волны 254 нм.
На хроматограмме испытуемого раствора, кроме основного пятна, допускается наличие дополнительных пятен анестезина и кислоты 4-аминобензойной, которые по величине и интенсивности поглощения не должны превышать пятна на хроматограммах раствора сравнения (b) и раствора сравнения (с), соответственно (не более 0.5 %).
Результаты анализа считаются достоверными, если выполняются требования теста «Проверка пригодности хроматографической системы».
Примечания. Приготовление раствора сравнения (а). 2.0 г СО ГФ РК или РСО прокаина гидрохлорида Р растворяют в воде Р и доводят объем полученного раствора тем же растворителем до 100.0 мл.
Приготовление раствора сравнения (b). 10.0 мг СО ГФ РК или РСО анестезина Р растворяют в 96 % спирте Р и доводят объем полученного раствора тем же растворителем до 100.0 мл.
ЭТАП
Приготовление раствора сравнения (с). 10.0 мг СО ГФ РК или РСО кислоты 4-аминобензойной Р растворяют в 96 % спирте Р и доводят объем полученного раствора тем же растворителем до 100.0 мл.
Проверка пригодности хроматографической системы. Хроматографическая система считается пригодной, если:
- на хроматограмме общей точки раствора сравнения (а), раствора сравнения (b) и раствора сравнения (с) четко делятся пятна новокаина (прокаина гидрохлорида), кислоты 4-аминобензойной и анестезина.
ЭТАП
+
1. R – NH2 + HCL + NaNO2 → [R – N ≡N] ∙ CL- + NaCL + H2O
+
[R – N ≡N] ∙ CL- + R – OH → R – N = N – R + HOH
Азокраситель
2. Провел реакцию идентификации на выделение бесцветного маслянистого осадка. Взял 0,2 г препарата, растворил в 2 мл воды, прибавил 0,5 мл раствора едкого натра; при этом выделяется бесцетный маслянистый соадок.
3. Определил наличие хлорида, для этого 0,02 г новокаина растворил в 2 мл воды, затем прибавил 0,5 мл разведенной кислоты азотной и 0,5 мл раствора нитрата серебра; при этом образовался белый творожистый осадок, который растворился в растворе аммиака.
HCL + AgNO3→ ↓AgCL + HNO3
AgCL + NH4OH→ [Ag (NH3)2] ∙ CL + HOH
Препарат дает характерную реакцию (а) на хлориды (ГФ РК I, т. 1, 2.3.1).
4. Определил, что соли диазония образуются в кислой среде, в связи с этим прибавляют к реакционной смеси щелочной раствор β – нафтола. Препарат дает характерную реакцию на первичные ароматические амины (ГФ РК I, т. 1, 2.3.1).
5. Правильно назвал, что азокраситель - оранжево-красного цвета осадок, хлорид серебра - белый творожистый осадок.
6. К 2 мл препарата прибавляют 0.2 мл кислоты серной разбавленной Р и 1 мл раствора калия перманганата Р; фиолетовое окрашивание раствора быстро исчезает (отличие от совкаина).
ЭТАП
1. Проведение методики «Количественное определение». 25.0 мл препарата помещают в коническую колбу вместимостью 250 мл, прибавляют 10 мл кислоты хлороводородной разбавленной Р, 3.0 г калия бромида Р (испытуемый раствор).
Испытуемый раствор охлаждают на ледяной бане и медленно титруют 0.05 М раствором натрия нитрита при постоянном перемешивании, прибавляя его сначала со скоростью 2 мл/мин, в конце титрования (за 0.5 мл до эквивалентного количества) - скорость титрования уменьшают до 0.05 мл /мин.
2. Молекулярная масса новокаина 272,77 г/моль.
3. В качестве индикатора используют раствор нейтрального красного (0.1 мл в начале и 0.1 мл в конце титрования), проводя титрование до перехода окрашивания раствора от красно-фиолетового до синего; или используют раствор тропеолина 00 в смеси с метиленовым синим (0.2 мл раствора тропеолина 00 и 0.1 мл раствора метиленового синего), проводя титрование до перехода окрашивания раствора от красно-фиолетового до голубого.
Параллельно проводят контрольный опыт.
4. 1 мл 0.05 М раствора натрия нитрита соответствует 0.01364 г C13H21ClN2O2 (новокаина).
5. Содержание новокаина в 1 мл препарата должно быть от 0.00475 г до 0.00525 г.
ЭТАП
1. Срок годности – период времени, в течение которого данное лекарственное средство по уровню качественных и количественных характеристик соответствует требованиям нормативной документации (НД).
2. Методы ускоренного старения в отличие от классических методов определения сроков годности позволяют за достаточно короткий период (15-115 дней) при 40-700С установить сроки хранения, что как правило совпадают с результатами, полученными при хранении лекарственных веществ при комнатной температуре в течение 3-5 лет. Оценку стабильности осуществляют, исследуя физические и химические изменения вещества.
1. Изотермический метод основан на применении правила Вант-Гоффа:
при повышении температуры на 100С скорость химической реакции возрастает в 2-4 раза.
2. Срок годности С при температуре хранения tхр связан с экспериментальным сроком годности Сэ при повышенной температуре экспериментального хранении tэ следующей зависимостью:
С = К ∙ Сэ,
tэ - tхр
где К = А 10
А = 2
Соответственно условиям лабораторного анализа, С = К ∙ Сэ + С0
Следовательно, экспериментальные сроки годности Сэ (I) и Сэ (II) найдены равными 243 и 70 суткам.
40 - 20
К = 2 10 = 4
60 - 20
К = 2 10 = 16
Сэ (I) = 243 сут ∙ 4 + 30 сут = 1002 сут (К = 4)
Сэ (II) = 70 ∙ 16 + 30 сут = 1150 сут (К = 16)
С = (1002 сут + 1150 сут): 2 = 1076 сут
5. Заключение. Оценку стабильности осуществил, исследуя химические изменения вещества: 1076 сут = 2 года 11 месяцев.
Пример из АНД
Упаковка. По 5 мл препарата в ампулы полиэтиленовые типа АП-5 по ТУ У 25.2-20390397-001:2007.
По 10 ампул вместе с инструкцией по медицинскому применению на государственном и русском языках помещают в пачку из картона для потребительской тары по ГОСТ 7933-89.
Групповая и транспортная тара в соответствии с ГОСТ 17768-90.
Маркировка. См. утвержденный макет упаковки.
Транспортирование. В соответствии с ГОСТ 17768-90.
Хранение. В оригинальной упаковке при температуре не выше 25 ˚С.
Срок хранения 2 года.
СПИРТ ЭТИЛОВЫЙ
1. Процесс получения этилового спирта из сырья, содержащего крахмал, заключается в том, что сырье измельчают и запаривают перегретым паром при 140-150 0С до образования густой массы в виде клейстера
2. Химические реакции получения спирта этилового:
3. 
4. 
5. 
6. Технологическая схема получения спирта этилового:
7. 
8. Спирт этиловый – это прозрачная, бесцветная, подвижная, летучая жидкость с характерным запахом и жгучим вкусом. Легко воспламеняется и горит голубым бездымным пламенем. Гигроскопичен.
9. Спирт смешивается во всех соотношениях с водой и большинством органических растворителей (эфиром, хлороформом).
ЭТАП
1. Не более 2.5х10-3 % (25 млн.-1, м/об). ГФ РК Т II, частная статья «Этанол (96 %, с. 581)».
2. 100 мл субстанции упаривают досуха на водяной бане и сушат при температуре 100-105°С) в течение 1 ч. Масса сухого остатка не должна превышать 2.5 мг. ГФ РК Т II, частная статья «Этанол (96 %, с. 581)».
3. Не более 10-4 % (1 млн.-1). Сухой остаток, полученный в испытании «Сухой остаток», растворяют в 1 мл 1 М кислоты хлороводородной, количественно переносят в мерную колбу вместимостью 100 мл, доводят объем раствора водой Р до 100 мл и перемешивают. 10 мл полученного раствора должны выдерживать испытание на железо.
4. Не более 2х10-4 % (2 млн.-1). 12 мл раствора, приготовленного для испытания на «Железо», должны выдерживать испытание на тяжелые металлы. Раствор сравнения готовят, используя 10 мл стандартного раствора свинца (2 млн.-1 Pb2+) Р.
ЭТАП
1. Идентификация проводится по двум методам: Первая идентификация: А, В (физико-химические методы) и Вторая идентификация: А, C, D (физико-химический метод и качественные реакции).
2. Первая идентификация: А, В
3. А. Субстанция должна соответствовать требованиям испытания «Относительная плотность» (2.2.5 ГФ РК, Т 1). От 0.805 до 0.812.
4. В. Инфракрасный спектр поглощения (абсорбционная спектрофотометрия в инфракрасной области, 2.2.24 ГФ РК, Т 1) субстанции должен соответствовать стандартному спектру СО ГФ РК этанола (96 %).
5. Вторая идентификация: А, C, D
6. А. См пункт 2
7. С. Химическая реакция с калием перманганатом и кислотой серной. На фильтровальной бумаге появляется интенсивное голубое окрашивание, бледнеющее через 10-15 минут.
8. D. Химическая реакция с раствором натрия гидроксида и раствором йода. Образуется желтый осадок.
9. Раствор 10 г/л калия перманганата Р, кислота серная разбавленная Р, натрия нитропруссид Р, пиперазина гидрат.
10. 0,1 мл субстанции смешивают в пробирке с 1 мл раствора 10 г/л калия перманганата Р и 0,2 мл кислоты серной разбавленной Р. Пробирку сразу накрывают фильтровальной бумагой, смоченной свежеприготовленным раствором, содержащим 0,1 г натрия нитропруссида Р и 0,5 г пиперазина гидрата Р в 5 мл воды Р; через несколько минут на бумаге появляется интенсивное голубое окрашивание, бледнеющее через 10-15 минут.
11. Раствор натрия гидроксида разбавленного Р, раствор йода.
12. К 0,5 мл субстанции прибавляют 5 мл воды Р, 2 мл раствора натрия гидроксида разбавленного Р, затем медленно добавляют 2 мл 0.05 М раствора йода; в течение 30 мин образуется желтый осадок.
ЭТАП
1. Ацетальдегид, метанол, ацетон, 2-пропанол, трет-бутанол, метилэтилкетон, 2-бутанол, циклогексан, бензол, 2-метил-1-пропанол, бутанол, ацеталь, метиизобутилкетон, пентанол, фурфурол, октанол.
2. Газовая хроматография с пламенно-ионизационным детектором.
3. Содержание суммы ацетальдегида и ацеталя в субстанции в пересчете на ацетальдегид не должно превышать 10-3 % (10 млн.-1, об/об).
4. Содержание бензола в субстанции не должно превышать 2·10-4 (2 млн-1, об/об).
5. На хроматограмме испытуемого раствора (б) сумма площадей всех пиков, кроме основного и пиков метанола, ацетальдегида, ацеталя и бензола, не должна превышать площадь пика 4-метилпентан-2-ола (0.03 % (300 млн-1, об/об).Не учитывают пики, площадь которых составляет менее 0.03 площади пика 4-метилпентан-2-ола на хроматограмме испытуемого раствора (б) (9·10-4 % (9 млн-1, об/об).
ЭТАП
1. Этиловый спирт разливают в специально оборудованные и предназначенные для него цистерны или резервуары, изготовленные из материалов, разрешенных уполномоченным органом в установленном порядке для контакта с этиловым спиртом. Цистерны и резервуары должны герметически закрываться крышками, иметь воздушники, оборудованные предохранительными клапанами. Для установления уровня спирта применяют поплавковые или другие безопасные указатели уровня. Цистерны и резервуары с этиловым спиртом, расположенные вне помещений, должны быть опломбированы.
Допускается разливать этиловый спирт-сырец в чистые бочки по ГОСТ 13950 или ГОСТ 6247, бутыли, канистры по ГОСТ 5105 или другие емкости, изготовленные из материалов, разрешенных уполномоченным органом в установленном порядке для контакта с продуктом данного вида, которые должны быть опечатаны или опломбированы. Упаковка и укупорка тары с этиловым спиртом-сырцом должны обеспечивать его сохранность и соответствовать требованиям ГОСТ 26319.
2. Транспортная маркировка - по ГОСТ 14192. Маркировка, характеризующая транспортную опасность груза, - по ГОСТ 19433 с указанием следующей информации:
- наименование предприятия-изготовителя, его адрес;
- наименование продукции;
- объем, дкл;
- масса брутто, кг;
- номер бочки, бутылки, канистры и партии;
- надпись «легковоспламеняющаяся жидкость»;
- знак опасности (чертеж 3), классификационный шифр 3212, номер ООН - 1170;
- обозначение настоящего стандарта.
3. Этиловый спирт транспортируют всеми видами транспорта в соответствии с правилами перевозки опасных грузов, действующими на данном виде транспорта, и правилами перевозки жидких грузов наливом в вагонах-цистернах, действующими на железнодорожном транспорте.
4. Хранение этилового спирта-сырца - в соответствии с инструкцией по приемке, хранению, отпуску, транспортированию и учету этилового спирта, утвержденной в установленном порядке. Спирт этиловый хранят в хорошо укупоренной таре, в прохладном месте, учитывая летучесть спирта, в защищенном от света месте. Срок хранения спирта не ограничен.
ТИМОЛ
1. Тимол может быть получен из м-крезола, который ацилируют, конденсируют с ацетоном и гидрируют.
2. Химическая реакция:
3. 
4. Технологическая схема получения тимола:
5. 
6. Тимол - крупные бесцветные кристаллы или кристаллический порошок с характерным запахом и пряно-жгучим вкусом, летуч с водяным паром.
7. Очень мало растворим в воде, очень легко растворим в 96 % спирте, легко растворим в эфирных и жирных маслах, умеренно растворим в глицерине.
ЭТАП
1. Прозрачность раствора (2.2.1). 1.0 субстанции растворяют в 10 мл раствора натрия гидроксида разбавленного Р. Опалесценция полученного раствора не должна превышать опалесценцию суспензии сравнения IV.
2. Цветность раствора (2.2.2. метод 2). Окраска раствора, приготовленного для испытания «Прозрачность раствора», не должна быть интенсивнее окраски раствора сравнения R6.
3. Кислотность. 1.0 г субстанции помещают в коническую колбу вместимостью 100 мл с притертой стеклянной пробкой, прибавляют 20 мл воды Р, кипятят до полного растворения, охлаждают, закрывают колбу и интенсивно встряхивают в течение 1 мин. Затем к полученному раствору добавляют несколько кристаллов субстанции для начала кристаллизации, интенсивно встряхивают в течение 1 мин и фильтруют. К 5 мл фильтрата добавляют 0.05 мл 0.01 М раствора натрия гидроксида; раствор должен быть желтым.
4. Родственные примеси. Определение проводят методом газовой хроматографии (2.2.28).
5. Сухой остаток. 2.00 г субстанции выпаривают на водяной бане и сушат при температуре от 100 0С до 105 0С в течение 1 ч. Масса сухого остатка не должна превышать 1.0 мг (0.05 %).
ЭТАП
1. Идентификация проводится по двум методам: Первая идентификация: В (физико-химические методы, качественная реакция) и Вторая идентификация: А, C, D (физико-химический метод и качественные реакции).
2. Первая идентификация: В
В. Инфракрасный спектр поглощения (2.2.24) субстанции, полученный в дисках, должен соответствовать спектру СО ГФ РК тимола.
3. Вторая идентификация: А, C, D
А. Температура плавления (2.2.14) от 48°С до 52 °С.
4. С. 0.2 г субстанции растворяют при нагревании в 2 мл раствора натрия гидроксида разбавленного Р, прибавляют 0.2 мл хлороформа Р и нагревают на водяной бане; появляется фиолетовое окрашивание.
D.Около 2 мг субстанции растворяют в 1 мл кислоты уксусной безводной Р, прибавляют 0.15 мл кислоты серной Р и 0.05 мл кислоты азотной Р; появляется сине-зеленое окрашивание.
С. 0.2 г субстанции растворяют при нагревании в 2 мл раствора натрия гидроксида разбавленного Р, прибавляют 0.2 мл хлороформа Р и нагревают на водяной бане; появляется фиолетовое окрашивание.
ЭТАП
1. Метод титрования.
0.500 г субстанции растворяют в 5.0 мл раствора 100 г/л натрия гидроксида Р и доводят объем раствора водой Р до 100.0 мл. 10.0 мл полученного раствора переносят в колбу с притертой пробкой, прибавляют 0,5 г калия бромида Р, 45 мл кислоты хлороводородной разбавленной Р, и интенсивно взбалтывая титруют 0,02 М раствором калия бромата до появления розового окрашивания, используя в качестве индикатора 0,3 мл раствора 1 г/л метилового оранжевого Р в начале титрования и 0,2 мл того раствора перед концом титрования.
2. Титрант: 0.02 М раствор калия бромата
Индикатор: 0.3 мл раствора 1 г/л метилового оранжевого Р.
Изменение окраски индикатора при рН 3,1-4,0.
Розовое окрашивание.
3. 1 мл 0,02 м раствора калия бромата соответствует 0,004506 г С10Н14О.
4. Расчет содержания лекарственного вещества в субстанции, в процентах (Х):
V·T·Kn·100
Х (%) = a,
где V – количество тированного раствора, израсходованного на титрование лекарственного вещества;
Т – титр, количество растворенного вещества, соответствующего 1 мл титрованного раствора, г.;
а – навеска порошка, взятая для анализа, г;
Kn–коэффициент поправки.
5. 1 мл 0,5 М раствора натрия гидроксида соответствует 45,04 мг С9Н8О4
ЭТАП
1. Лекарственное вещество выпускается в форме таблеток, таблеток шипучих, таблеток, кишечнорастворимых для приема внутрь.
2. По 10 таблеток в контурную ячейковую упаковку из пленки поливинилхлоридной по ГОСТ 25250-88 и фольги алюминиевой по ГОСТ 745-2003.
По N контурных упаковки вместе с инструкцией по медицинскому применению на государственном и русском языках помещают в коробку из картона по ГОСТ 7933-89.
3. Маркировка упаковки на государственном и русском языках в соответствии с Техническим регламентом «Требования к безопасности лекарственных средств», утверждено постановлением Правительства Республики Казахстан от 14 июля 2010 года № 712.
4. Препарат применяется в качестве антисептического и противоглистного средства. Хранят в защищенном от света месте.
Date: 2016-07-18; view: 2509; Нарушение авторских прав