Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Физическая Химия
|
|
Г О С К И Н О Р О С С И И
САНКТ-ПЕТЕРБУРГСКИЙ ГОСУДАРСТВЕННЫЙ
УНИВЕРСИТЕТ КИНО И ТЕЛЕВИДЕНИЯ
Кафедра общей, органической и физической химии
ФИЗИЧЕСКАЯ ХИМИЯ
Методические указания по выполнению лабораторных
работ для студентов специальности 240504
"Технология кинофотоматериалов и магнитных носителей"
дневного и заочного отделений
Часть III. Термохимия и ионные равновесия
Санкт-Петербург
Составитель: проф. Л.Л. Кузнецов
Рецензент: В.В. Митрофанов, доцент кафедры общей химической технологии и промышленной экологии
Рекомендовано к изданию в качестве методических указаний по лабораторному практикуму по физической химии кафедрой общей, органической и физической химии.
Протокол заседания кафедры общей, органической и физической химии
№ 8 от 20 мая 2009 г.
ã СПбГУКиТ, 2009 г.
РАБОТА № 5
ФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ТЕРМОДИНАМИЧЕСКОЙ КОНСТАНТЫ КИСЛОТНОЙ ДИССОЦИАЦИИ ОДНОЦВЕТНОГО ИНДИКАТОРА
Цель работы − определение константы кислотной диссоциации слабой кислоты, являющейся одноцветным индикатором. Определение проводится фотометрическим методом, основанным на пропорциональности между оптической плотностью раствора индикатора и концентрацией его аниона. По величине оптической плотности определяют степень диссоциации слабой кислоты при определенном значении рН. Это позволяет вычислить концентрационную и термодинамическую константы ее диссоциации.
1. Теоретическая часть
1.1. Кислоты и основания.
Согласно теории Бренстеда и Лоури кислота - это вещество, способное отдавать протон, а основание - вещество, способное присоединять протон, например:
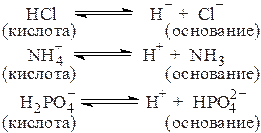
и т.д.
Поскольку в водных растворах, органических растворителях и их смесях протоны в свободном виде практически не существуют, любое кислотно-основное равновесие в растворе может иметь место лишь в случае наличия как кислоты, отдающей протон, так и основания, этот протон принимающего, например:
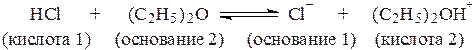 .
.
При этом кислота, потеряв протон, превращается в сопряженное с ним основание, а основание, присоединившее протон, – в сопряженную кислоту. Часто в роли кислоты или основания выступают молекулы растворителя. Если растворитель присоединяет протон, проявляя свойства основания, он называется протофильным. Такими растворителями являются вода, спирты, кетоны, эфиры, аммиак и различные амины, карбоновые кислоты. Все эти вещества способны присоединять протоны, благодаря наличию в их молекулах атомов азота или кислорода, имеющих неподеленные электронные пары (заполненные несвязывающие молекулярные орбитали):
 .
.
В том случае, когда растворитель отдает протоны, играя роль кислоты, он называется протогенным. Типичными примерами являются вода, спирты, карбоновые и минеральные кислоты, жидкий хлороводород и т.д. Растворители, способные как принимать, так и отдавать протоны, называются амфипротонными (вода, спирты, карбоновые кислоты). Растворители, не способные ни принимать, ни отдавать протоны, называются апротонными. Примерами таких растворителей могут служить, например, хлороформ, бензол, гексан и т.д. В этих растворителях кислотно-основные равновесия не устанавливаются.
В зависимости от свойств растворителя, одно и то же вещество, растворенное в нем, может проявлять либо кислотные, либо основные свойства. Так, карбоновые кислоты в воде проявляют свойства кислот, а в серной кислоте - свойства оснований, например:
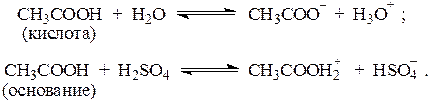
В дальнейшем будут рассматриваться лишь водные растворы кислот и оснований.
1.2. Кислотность водных растворов кислот и оснований.
Вода является амфипротонным растворителем, в котором даже в отсутствие посторонних веществ устанавливается равновесие:
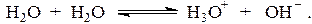
Термодинамическая константа этого равновесия имеет вид:
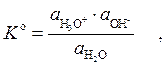

где 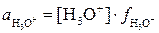 ;
; 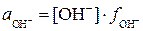 - активности ионов, равные произведению концентрации на коэффициент активности. Поскольку концентрации образующихся ионов очень малы, коэффициенты активности близки к единице (
- активности ионов, равные произведению концентрации на коэффициент активности. Поскольку концентрации образующихся ионов очень малы, коэффициенты активности близки к единице (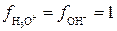 ) и поэтому активности ионов практически равны их концентрациям:
) и поэтому активности ионов практически равны их концентрациям: 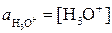 ;
; 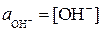 . Концентрацию, а значит и активность недиссоциированных молекул воды можно считать при данных условиях величинами постоянными и не зависящими от степени диссоциации (при комнатной температуре
. Концентрацию, а значит и активность недиссоциированных молекул воды можно считать при данных условиях величинами постоянными и не зависящими от степени диссоциации (при комнатной температуре 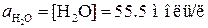 ). Произведение константы равновесия на активность воды
). Произведение константы равновесия на активность воды 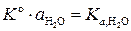 называется термодинамической константой диссоциации воды:
называется термодинамической константой диссоциации воды:
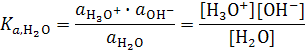 .
.
В свою очередь, произведение константы диссоциации на активность воды называется ионным произведением воды:
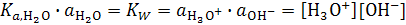 .
.
При 20°С величина 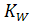 составляет 0.86×10-14 моль2/л2. Поскольку
составляет 0.86×10-14 моль2/л2. Поскольку 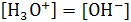 , концентрация, следовательно и активность ионов гидроксония в воде составляют:
, концентрация, следовательно и активность ионов гидроксония в воде составляют: 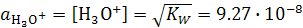 моль/л. Любой водный раствор с таким содержанием ионов
моль/л. Любой водный раствор с таким содержанием ионов 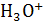 называется нейтральным. Если их концентрация в растворе больше, раствор называют кислым, если меньше - щелочным. При добавлении к воде кислоты увеличивают концентрацию ионов гидроксония за счет реакции с водой, а основания - уменьшают, поскольку реагируют с водой и ионами гидроксония:
называется нейтральным. Если их концентрация в растворе больше, раствор называют кислым, если меньше - щелочным. При добавлении к воде кислоты увеличивают концентрацию ионов гидроксония за счет реакции с водой, а основания - уменьшают, поскольку реагируют с водой и ионами гидроксония:
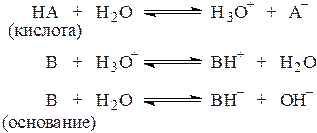
Кислотность раствора, определяемая концентрацией или активностью ионов , численно характеризуется величиной рН, или водородным показателем, равным отрицательному логарифму активности катионов гидроксония:
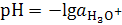 .
.
Для нейтрального раствора: 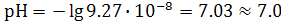 . Кислые растворы имеют рН меньше семи, щелочные - больше семи. При известном значении рН активность ионов гидроксония легко находится как
. Кислые растворы имеют рН меньше семи, щелочные - больше семи. При известном значении рН активность ионов гидроксония легко находится как 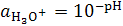 .
.
1.3. Сила кислот и оснований в водных растворах.
Сила кислоты в водном растворе тем больше, чем дальше равновесие
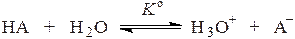
смещено вправо. Количественно сила кислоты характеризуется величиной термодинамической константы этого равновесия:
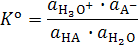 .
.
Для достаточно разбавленных растворов кислот активность воды не зависит от степени диссоциации кислоты и является величиной постоянной и равной 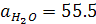 моль/л. Произведение термодинамической константы этого равновесия на активность воды называется термодинамической константой диссоциации кислоты:
моль/л. Произведение термодинамической константы этого равновесия на активность воды называется термодинамической константой диссоциации кислоты:
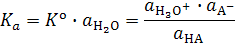 .
.
Чем больше эта величина, тем в большей степени кислота диссоциирует на ионы, тем больше сила кислоты.
Для характеристики силы основания применяют величину термодинамической константы диссоциации его сопряженной кислоты:
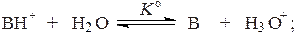
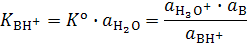 .
.
Чем меньше эта величина, то есть чем дальше это равновесие смещено влево, тем в большей степени основание B протонируется с образованием его сопряженной кислоты, тем выше сила основания (тем слабее его сопряженная кислота).
На практике пользуются не самими константами, а их отрицательными логарифмами. Логарифмирование приведенных выше уравнений дает:
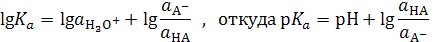 (1)
(1)
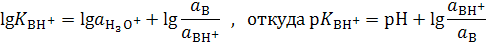 , (2)
, (2)
 где p Ka = −lg Ka, a p K BH+ = −lgKBH+. Анализ выражений (1) и (2) показывает, что p Ka и p K BH+равны величинам рН, при которых кислота или основание ионизированы наполовину (при a A- = a HAи a B= a BH+ соответственно). Так, для синильной кислоты (HCN) величина p Ka = 9.31, а для муравьиной (HCOOH) p Ka = 3.75. Следовательно, для диссоциации синильной кислоты на 50% необходима щелочная среда (рН 9.31), в то время как намного более сильная муравьиная кислота диссоциирует наполовину даже в кислой среде (рН 3.75). Аналогично для анилина C6H5NH2 p K BH+ = 4.63, а для аммиака NH3 p K BH+ = 9.25. Это означает, что слабое основание анилин превращается на 50% в соль лишь в кислой среде (рН 4.63), а значительно более сильное основание аммиак наполовину превращается в ион аммония даже в весьма щелочной среде (рН 9.25). Следовательно, чем меньше величина p Ka, тем сила кислоты больше, а чем меньше p K BH+, тем сила основания меньше.
где p Ka = −lg Ka, a p K BH+ = −lgKBH+. Анализ выражений (1) и (2) показывает, что p Ka и p K BH+равны величинам рН, при которых кислота или основание ионизированы наполовину (при a A- = a HAи a B= a BH+ соответственно). Так, для синильной кислоты (HCN) величина p Ka = 9.31, а для муравьиной (HCOOH) p Ka = 3.75. Следовательно, для диссоциации синильной кислоты на 50% необходима щелочная среда (рН 9.31), в то время как намного более сильная муравьиная кислота диссоциирует наполовину даже в кислой среде (рН 3.75). Аналогично для анилина C6H5NH2 p K BH+ = 4.63, а для аммиака NH3 p K BH+ = 9.25. Это означает, что слабое основание анилин превращается на 50% в соль лишь в кислой среде (рН 4.63), а значительно более сильное основание аммиак наполовину превращается в ион аммония даже в весьма щелочной среде (рН 9.25). Следовательно, чем меньше величина p Ka, тем сила кислоты больше, а чем меньше p K BH+, тем сила основания меньше.
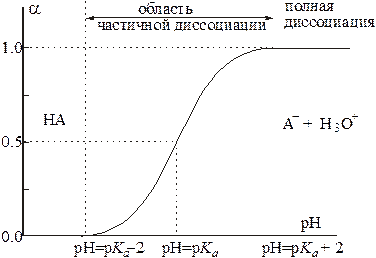
Рис. 1.Зависимость степени диссоциации слабой кислоты от рН среды.
Графически зависимость степени диссоциации кислоты НА от величины рН среды представлена на рис.1. Зависимость степени протонирования основания от рН среды имеет вид прямо противоположный представленному на рис.1. При pH = p K BH+ − 2 основание полностью протонировано и находится в растворе в виде 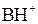 , при pH = p K BH+ основание протонировано наполовину (50% его находится в виде свободного основания В и 50% в виде соли
, при pH = p K BH+ основание протонировано наполовину (50% его находится в виде свободного основания В и 50% в виде соли 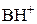 ) и при pH = p K BH+ + 2 основание практически не протонировано. Степень диссоциации кислоты или протонирования основания в среде с данным значением рН легко вычислить по уравнению (1) или (2) соответственно.
) и при pH = p K BH+ + 2 основание практически не протонировано. Степень диссоциации кислоты или протонирования основания в среде с данным значением рН легко вычислить по уравнению (1) или (2) соответственно.
1.4. Буферные растворы.
Добавление сильной кислоты или сильного основания к воде или водному раствору электролита вызывает изменение рН. Буферной емкостью раствора (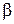 ) называется количество г-эквивалентов щелочи или сильной кислоты, необходимое для изменения рН одного литра раствора на единицу:
) называется количество г-эквивалентов щелочи или сильной кислоты, необходимое для изменения рН одного литра раствора на единицу:
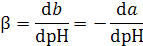 ,
,
где b - количество г-эквивалентов добавленной щелочи; а - количество г-эквивалентов добавленной кислоты. Буферная емкость всегда больше нуля. Это котангенс угла наклона касательной, проведенной к зависимости рН от количества г-эквивалентов добавленной кислоты или щелочи. Поэтому для того, чтобы проследить зависимость буферной емкости раствора от его состава и кислотности среды, необходимо рассмотреть изменение величины рН раствора при добавлении к нему сильной кислоты или щелочи.
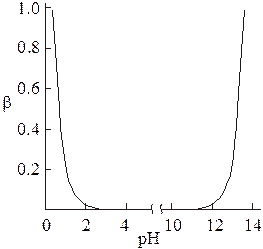 Добавление кислоты или щелочи к дистиллированной воде довольно сильно меняет ее кислотность в интервале рН от ~3 до ~11 и сравнительно слабо в более кислой и в более щелочной среде. Таким образом, вода обладает относительно высокой буферной емкостью лишь в сильно кислых и в сильно щелочных средах (рис.2).
Добавление кислоты или щелочи к дистиллированной воде довольно сильно меняет ее кислотность в интервале рН от ~3 до ~11 и сравнительно слабо в более кислой и в более щелочной среде. Таким образом, вода обладает относительно высокой буферной емкостью лишь в сильно кислых и в сильно щелочных средах (рис.2).
Рис.2. Зависимость буферной емкости воды от величины рН среды.
Напротив, добавление сильной кислоты к раствору соли слабой кислоты, например к раствору ацетата натрия, слабо меняет рН среды в довольно широком интервале кислотности среды. Аналогично меняется рН и водного раствора слабой кислоты, например уксусной, при добавлении к нему щелочи (рис.3, а). Следовательно, такие растворы обладают высокой буферной емкостью не только в сильно кислых или щелочных средах. В общем виде буферная емкость раствора слабой кислоты зависит от ее концентрации (С), силы кислоты (величины ее Ka) и от кислотности среды (величины рН, то есть от 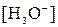 ) (3):
) (3):
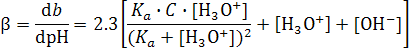 . (3)
. (3)
В графическом виде эта зависимость представлена на рис.3, б. Из него и анализа зависимости (3) следует, что в растворе, содержащем слабую кислоту и ее соль, при рН меньше ~3 и больше ~11 буферная емкость растет, как и в случае чистой воды с увеличением кислотности и щелочности среды:
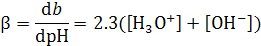 .
.
В среде с промежуточной кислотностью (рН между ~3 и ~11) буферная емкость раствора определяется выражением (4)
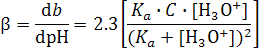 . (4)
. (4)
а при Ka = [H3O+], то есть при рН = p Ka буферная емкость раствора максимальна и прямо пропорциональна концентрации слабой кислоты. Действительно, из (4) следует, что при Ka = [H3O+]
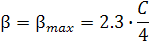 .
.
Это свойство водных растворов, содержащих слабую кислоту и ее соль, используется для приготовления так называемых буферных растворов, способных в заметной степени поддерживать примерно постоянное значение рН при добавлении небольших количеств сильных кислот или щелочей. Такие растворы имеют достаточно высокую буферную емкость в интервале от рН = p Ka - 1 до рН = p Ka + 1. По этой причине для приготовления буферных растворов необходимо подбирать соответствующую слабую кислоту, p Ka которой возможно ближе к требуемой величине рН.
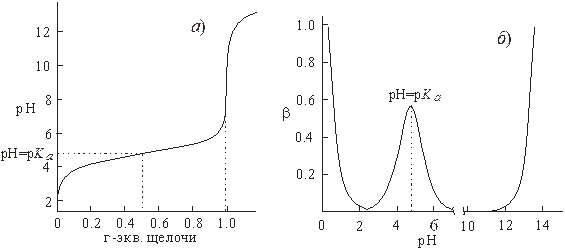
Рис.3. Зависимость рН 1 литра 1 молярного водного раствора уксусной кислоты от количества добавленной щелочи (а). Зависимость буферной емкости водного раствора уксусной кислоты и ацетата натрия от величины рН среды (б).
Величина рН буферного раствора зависит от концентрации в нем слабой кислоты и ее соли. Поскольку концентрации компонентов буферного раствора достаточно высоки и a A- = [A−]· f A−, а a HA = [HA]· f HA, из (1) следует:
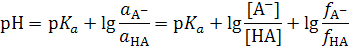 .
.
Соль слабой кислоты является сильным электролитом, диссоциирующим в водных растворах нацело, поэтому концентрация анионов равна концентрации соли: [A−] = [соль](диссоциацией слабой кислоты можно пренебречь). В то же время, поскольку слабая кислота диссоциирована лишь в очень малой степени, то [HA] = [кислота] и значит:
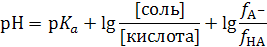 .
.
Следовательно, рН буферного раствора зависит от соотношения концентраций слабой кислоты и ее соли. Буферный раствор тем кислее, чем выше концентрация кислоты и наоборот. При равных концентрациях кислоты и соли, то есть если [соль] = [кислота], рН раствора практически равен величине p Ka слабой кислоты:
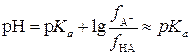 .
.
Разбавление буферного раствора водой очень мало влияет на величину его рН, поскольку оно не изменяет отношение [соль] / [кислота] и лишь в незначительной степени изменяет отношение коэффициентов активности.
1.5. Фотоколориметрический метод анализа.
Фотоколориметрическим называется метод анализа, основанный на измерении поглощения света веществом. В самом общем виде энергия одной молекулы за вычетом кинетической энергии ее поступательного движения складывается из вращательной энергии молекулы, колебательной энергии составляющих ее атомов, энергии электронов, входящих в электронные оболочки атомов и энергии, заключенной в ядрах атомов:
ε = ε вращат. + ε колеб. + ε электр. + ε ядер .
Взаимодействие квантов электромагнитного излучения с молекулами вещества приводит к изменению их энергетического состояния (энергия ядер при этом остается постоянной и поэтому Δ ε ядер = 0):
Δ ε = Δ ε вращат. + Δ ε колеб. + Δ ε электр..
Изменение энергии одного моля таких молекул в NA = 6.02×1023 раз больше и также является суммой:
Δ E = Δ ε · NA = Δ E вращат. + Δ E колеб. + Δ E электр..
Вращательные уровни энергии молекул расположены достаточно близко друг к другу, поэтому величина Δ E вращат. мала и составляет менее ~0.5 кДж/моль. Величины Δ E колеб. и Δ E электр.значительно больше и составляют ~6 - 8 кДж/моль и ~120 - 800 кДж/моль, соответственно.
Поглощение энергии квантов микроволнового излучения с длиной волны более ~300000 нм и энергией не более ~0.5 кДж/моль увеличивает лишь энергию вращательного движения молекул. В соответствии с условием резонанса поглощаются только те кванты, энергия которых удовлетворяет условию
hν = Δ E.
При этом возникают вращательные спектры поглощения молекул.
Энергии квантов инфракрасного излучения достаточно для изменения как вращательной, так и колебательной энергии молекул, поэтому при облучении вещества таким излучением возникают вращательно-колебательные, или ИК-спектры.
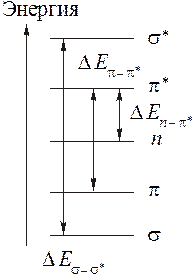 Кванты видимого света (длина волны от ~400 до ~760 нм) и ультрафиолетового излучения (длина волны менее ~400 нм) имеют энергию достаточную для увеличения энергии электронных оболочек молекул. В самом общем виде энергетические уровни электронов в молекуле можно представить следующим образом.
Кванты видимого света (длина волны от ~400 до ~760 нм) и ультрафиолетового излучения (длина волны менее ~400 нм) имеют энергию достаточную для увеличения энергии электронных оболочек молекул. В самом общем виде энергетические уровни электронов в молекуле можно представить следующим образом.
Если внешние электроны в молекуле находятся только на связывающих s-молекулярных орбиталях, как в молекулах насыщенных углеводородов, при поглощении энергии происходит переход одного из них на разрыхляющую s*-молекулярную орбиталь. Величина Δ E для s − s* перехода довольно велика, поэтому в данном случае поглощаются кванты только с очень большой энергией. Такой энергии соответствует жесткое ультрафиолетовое излучение с высокой частотой и малой длиной волны (λмакс. < 150×10-9 м, или 150 нм), практически недоступной для серийных спектрометров. Если в молекуле вещества имеются группировки с заполненными связывающими  -молекулярными орбиталями, такие как C=C, N=N, N=C, C=O, C=S и т.д., при поглощении квантов электромагнитного излучения происходит переход одного из -электронов на разрыхляющую
-молекулярными орбиталями, такие как C=C, N=N, N=C, C=O, C=S и т.д., при поглощении квантов электромагнитного излучения происходит переход одного из -электронов на разрыхляющую  -молекулярную орбиталь. Величина Δ E в этом случаезначительно меньше чем для s − s* перехода, поэтому такие молекулы поглощают излучение с большей длиной волны, доступной для измерений, "видимой" для спектрометров. Такие группировки называются хромофорами (дословно - придающими окраску).
-молекулярную орбиталь. Величина Δ E в этом случаезначительно меньше чем для s − s* перехода, поэтому такие молекулы поглощают излучение с большей длиной волны, доступной для измерений, "видимой" для спектрометров. Такие группировки называются хромофорами (дословно - придающими окраску).
| Формула | λмакс., нм | Формула | λмакс., нм |
| CH2=CH2 | 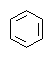
| ||
| CH2=CH-CH=CH2 | 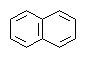
| ||
| CH2=CH-CH=CH-CH=CH2 | 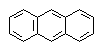
|
Чем больше в молекуле сопряженных друг с другом хромофоров, тем меньше величина Δ E, тем больше длина волны поглощаемого излучения.
Если кроме хромофорных групп в молекуле имеются сопряженные с ними группировки, содержащие заполненные электронами несвязывающие n -молекулярные орбитали (свободные электронные пары), появляется возможность для n - перехода. Поскольку Δ E для него довольно мала, то длина волны поглощаемого излучения в таких молекулах значительно увеличивается. Группировки, сдвигающие поглощение в длинноволновую область, называются ауксохромами. Типичными ауксохромамиявляются атомы азота, входящие в аминогруппы, спиртовые и эфирные атомы кислорода, атомы галогенов и т.д.:
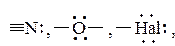
то есть группировки -NH2, -OH, -O-R, -Cl и другие. Типичным примером влияния ауксохрома на спектр поглощения молекулы является введение гидроксильной группы в молекулу нитробензола. Под влиянием ауксохрома -OH полоса поглощения нитробензола смещается в длинноволновую область от 250 нм для нитробензола до 320 нм для п- нитрофенола:
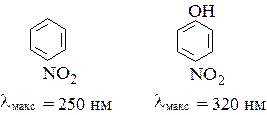 .
.
Согласно закону светопоглощения Бугера-Ламберта-Бера степень уменьшения интенсивности света пропорциональна числу поглощающих свет частиц. Если выделить элементарный объем с площадью S= 1 см2 и толщиной d l см, то входящий в него поток света с интенсивностью I Дж/см2 уменьшится на величину d I:
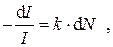
где d N- число частиц в элементарном объеме d V = S× d l. Так как S= 1 см2, то d V = d l см3. Если концентрация поглощающих свет частиц равна C моль/л, то величина d N = C×NA× d V× 10-3 частиц (N A = 6.02×1023 моль-1 - число Авогадро)и значит
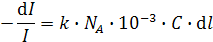 .
.
При прохождении света через слой среды толщиной l его интенсивность уменьшится от I oдо I. После интегрирования от I o до I и от 0 до l можно получить
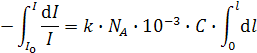
 ; или
; или 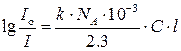 .
.
Если обозначить 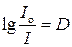 - оптическая плотность среды, в логарифмическом масштабе показывающая во сколько раз уменьшилась интенсивность света после прохождения через среду, а
- оптическая плотность среды, в логарифмическом масштабе показывающая во сколько раз уменьшилась интенсивность света после прохождения через среду, а 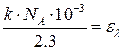 - молярный коэффициент экстинкции, численно равный оптической плотности среды с толщиной слоя 1 см и концентрацией частиц 1 моль/л, то закон светопоглощения можно записать как
- молярный коэффициент экстинкции, численно равный оптической плотности среды с толщиной слоя 1 см и концентрацией частиц 1 моль/л, то закон светопоглощения можно записать как
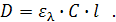
Оптическая плотность среды пропорциональна мольной концентрации частиц поглощающих свет и толщине поглощающего слоя. Величина молярного коэффициента экстинкции  зависит от свойств частиц и длины волны света. Зависимость коэффициента экстинкции от длины волны является характеристикой данного вещества и называется его электронным спектром поглощения.
зависит от свойств частиц и длины волны света. Зависимость коэффициента экстинкции от длины волны является характеристикой данного вещества и называется его электронным спектром поглощения.
При постоянной толщине поглощающего слоя (l)оптическая плотность D раствора вещества при данной длине волны пропорциональна его концентрации. График зависимости D от C называется калибровочным. Его строят, измеряя оптические плотности растворов с известной концентрацией вещества. Чаще всего график является линейным лишь в достаточно разбавленных растворах, в которых закон Бугера-Ламберта-Бера выполняется точно (ε = const). При относительно больших концентрациях вещества молярный коэффициент экстинкции не остается постоянным и линейность графика нарушается. Калибровочные графики используют для определения концентрации вещества (Cx) в растворе путем измерения его оптической плотности (Dx). В области выполнения закона Бугера-Ламберта-Бера более удобным оказывается не графическое определение концентрации, а ее вычисление с использованием молярного коэффициента экстинкции, определяемого как тангенс угла наклона линейной части калибровочного графика:
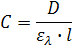 .
.
Для повышения чувствительности фотоколориметрического метода и увеличения его точности измерения проводят при длине волны, соответствующей максимальному значению коэффициента экстинкции.
2. Определение термодинамической константы кислотной диссоциации одноцветного индикатора
Вещества, поглощающие свет в видимой области спектра с длиной волны 400 нм и более, являются окрашенными. Если окраска вещества изменяется под влиянием каких-либо факторов (температура, кислотность среды и т.п.), оно называется соответствующим индикатором (индикатором температуры, индикатором кислотности и т.д.). Индикаторами кислотности чаще всего являются слабые кислоты или основания, степень ионизации которых зависит от кислотности среды.
Одноцветным индикатором называется слабая кислота или слабое основание, которое при изменении рН среды либо приобретает, либо теряет свою окраску. Это происходит потому, что либо нейтральная молекула, либо ион, образующийся при ее ионизации поглощает свет в видимой области спектра. Двухцветный индикатор при изменении рН среды изменяет свою окраску. В этом случае и нейтральная молекула и ион поглощают свет в видимой области спектра, однако их спектры поглощения различны.
Так, например, слабая органическая кислота - п -нитрофенол практически бесцветна, поскольку ее спектр поглощения имеет максимум в области ближнего ультрафиолета (λмакс.< 400 нм), а в видимой области она свет не поглощает. При диссоциации п -нитрофенола происходит образование п -нитрофенолят-аниона, в котором с p-электронной системой фенильного ядра сопряжена не одна, а две электронные пары атома кислорода. В результате взаимодействия такого ауксохрома с сопряженной системой хромофоров происходит значительный сдвиг длины волны поглощаемого излучения от 320 нм для п -нитрофенола до 400 нм для п -нитрофенолят-аниона и раствор из бесцветного становится желтым.
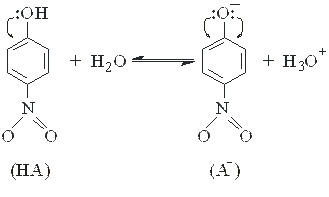
Если равновесие диссоциации слабой кислоты записать как
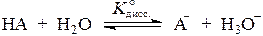 ,
,
термодинамическая константа этого равновесия принимает вид:
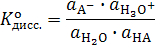 ,
,
а термодинамическая константа кислотности слабой кислоты равная произведению константы равновесия на активность воды принимает вид:
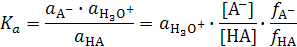 .
.
Коэффициент активности нейтральных молекул HA зависит только от их концентрации и, поскольку для определения Ka используют достаточно низкие концентрации индикатора (10-3 - 10-4 моль/л), величина f HA близка к единице. Коэффициент активности анионов индикатора зависит от концентрации любых ионов, находящихся в растворе, то есть от ионной силы раствора. Согласно первому приближению теории сильных электролитов Дебая-Хюккеля средний коэффициент активности иона в растворе определяется выражением
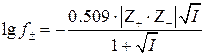 , (5)
, (5)
где 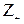 и
и 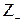 - заряд катиона и аниона, образующихся при диссоциации слабой кислоты; I - ионная сила раствора, определяемая как полусумма произведений концентраций ионов на квадраты их зарядов:
- заряд катиона и аниона, образующихся при диссоциации слабой кислоты; I - ионная сила раствора, определяемая как полусумма произведений концентраций ионов на квадраты их зарядов: 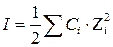 .
.
Следовательно, для вычисления термодинамической константы кислотности слабой кислоты достаточно определить активность ионов гидроксонияи отношение концентраций аниона и недиссоциированной формы кислоты. Поскольку концентрация аниона равна произведению степени диссоциации на общую концентрацию кислоты: [A-] = a× C HA, а концентрация недиссоциированной формы равна разности общей концентрации и концентрации ее диссоциированной формы
[HA] = C HA - a× C HA = C HA (1-a), то
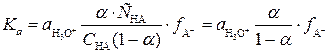 ,
,
или после логарифмирования: 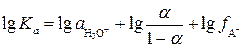 .
.
Поскольку 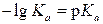 , а
, а 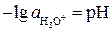 , то
, то
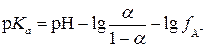 .
.
Степень диссоциации слабой кислоты при данном значении рН легко может быть найдена фотоколориметрическим методом в том случае, если слабая кислота является одноцветным индикатором, незаряженная форма которого является бесцветной, а анион - окрашен. Так, на рис. 2а приведен спектр поглощения раствора п -нитрофенола в кислой среде (рН < 5), когда равновесие его диссоциации полностью сдвинуто влево. В этом случае a = 0, а значит п -нитрофенол находится в растворе в виде недиссоциированной формы НА. Спектр раствора представляет собой спектр недиссоциированной формы. В достаточно щелочной среде (рН > 9) степень диссоциации п -нитрофенола равна единице и он находится в растворе исключительно в виде анионов А-. Спектр раствора представляет собой спектр поглощения аниона. Если рН раствора достаточен лишь для частичной диссоциации п -нитрофенола, спектр поглощения представляет собой сумму спектров аниона и недиссоциированной формы. Оптическая плотность раствора п -нитрофенола, измеренная при l = 400 нм в условиях, когда молярный коэффициент экстинкции недиссоциированной формы НА близок к нулю, определяется лишь поглощением аниона 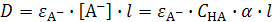 . При данной концентрации п -нитрофенола зависимость оптической плотности от рН (рис. 2 б)отражает зависимость величины степени его диссоциации от рН, сходную с представленной на рис.1.
. При данной концентрации п -нитрофенола зависимость оптической плотности от рН (рис. 2 б)отражает зависимость величины степени его диссоциации от рН, сходную с представленной на рис.1.
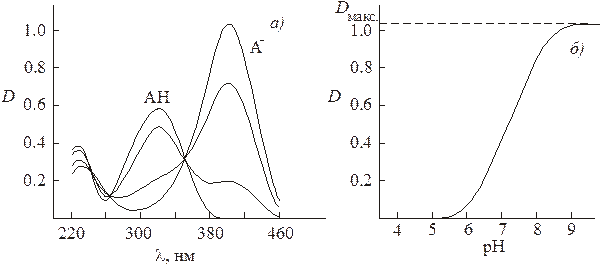
Рис.2. Спектр поглощения раствора п -нитрофенола при различных рН среды (а). Зависимость оптической плотности раствора п -нитрофенола от рН среды при l = 400 нм (б).
В соответствии с изменением степени диссоциации, с ростом рН раствора оптическая плотность меняется от 0 до 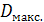 . Степень диссоциации при некотором данном значении рН легко может быть найдена как отношение
. Степень диссоциации при некотором данном значении рН легко может быть найдена как отношение 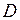 к , поскольку
к , поскольку
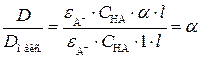 .
.
Следовательно, для точного определения термодинамической константы кислотной диссоциации одноцветного индикатора достаточно приготовить его растворы с постоянной концентрацией С HAи такими значениями рН среды, при которых он диссоциирован лишь частично, то есть в интервале рН» p Ka ± 1. Измерение оптических плотностей таких растворов позволяет определить степень диссоциации и вычислить константу диссоциации. Для определения интервала значений рН таких растворов сначала находят приблизительное значение 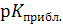 и только после этого находят несколько точных значений, из которых определяют значения констант кислотности (Ka =
и только после этого находят несколько точных значений, из которых определяют значения констант кислотности (Ka = 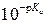 ). Среднее арифметическое из них является точным значением термодинамической константы диссоциации одноцветного индикатора.
). Среднее арифметическое из них является точным значением термодинамической константы диссоциации одноцветного индикатора.
3. Ход работы и обработка полученных данных
3.1. В мерную колбу емкостью 50 мл с помощью пипетки помещают 5 мл раствора индикатора, доливают до метки 0.02 н водный раствор NaOH, переливают в коническую колбу и тщательно перемешивают.
3.2. Полученный раствор заливают в кювету фотоколориметра, предварительно ополоснув ее тем же раствором, и измеряют величину (измерение проводят не менее трех раз, усреднив полученные результаты).
3.3. Готовят раствор индикатора с рН 7 для о- и п- нитрофенола и с рН 8 для м- нитрофенола. Для этого в мерную колбу емкостью 50 мл с помощью пипетки заливают 5 мл раствора индикатора и добавляют компоненты буферного раствора в количестве, определяемом из табл.2. Полученный раствор тщательно перемешивают.
3.4. Измеряют величину 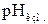 (см. инструкцию к рН-метру) и с помощью фотоколориметра измеряют величину оптической плотности полученного раствора.
(см. инструкцию к рН-метру) и с помощью фотоколориметра измеряют величину оптической плотности полученного раствора.
3.5. Используя значения , и , вычисляют степень диссоциации a и затем приближенное значение р К индикатора: 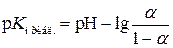 .
.
3.6. Вычисляют расчетные значения рН семи буферных растворов, лежащих в пределах 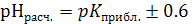 :
: 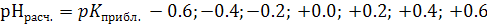 .
.
3.7. Как описано выше (п. 3.3) готовят растворы индикатора в буферных растворах с величинами 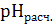 (требуемые объемы компонентов буферного раствора находят из табл. 2), измеряют для них величины
(требуемые объемы компонентов буферного раствора находят из табл. 2), измеряют для них величины 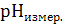 и значения оптической плотности D (как описано в п. 3.2). Заносят полученные данные в табл. 1.
и значения оптической плотности D (как описано в п. 3.2). Заносят полученные данные в табл. 1.
Таблица 1
Индикатор: (название) =; 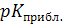 =.
=.
| № р-ра |
|
| Di | 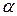
| 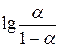
| p Ki | Ki |
3.8. Пользуясь величинами D, D макс. и pHизмер. вычисляют 7 отдельных значений степени диссоциации 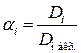 , значений
, значений 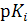 по формуле
по формуле 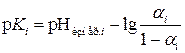 и, наконец, значений концентрационных констант
и, наконец, значений концентрационных констант 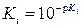 .
.
3.9. Находят из них среднее арифметическое (K средн.), затем по уравнению (5) вычисляют значение коэффициента активности аниона индикатора (ионная сила раствора составляет 0.1) и используют его для вычисления термодинамической константы кислотности 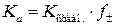 и величины
и величины 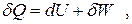 .
.
Таблица 2
Буферная система (ионная сила составляет 0.1):
0.1 н раствор КН2PO4 +0.05 н раствор буры (Na2B4O7)
| рН* | 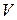 р-ра
КН2PO4, мл р-ра
КН2PO4, мл
| рН* | р-ра
КН2PO4, мл
| рН* | р-ра
КН2PO4, мл
|
| 6.0 | 39.5 | 7.1 | 27.1 | 8.2 | 19.6 |
| 6.1 | 38.4 | 7.2 | 26.1 | 8.3 | 18.3 |
| 6.2 | 37.4 | 7.3 | 25.4 | 8.4 | 17.1 |
| 6.3 | 36.3 | 7.4 | 24.7 | 8.5 | 15.9 |
| 6.4 | 35.2 | 7.5 | 24.1 | 8.6 | 14.6 |
| 6.5 | 34.1 | 7.6 | 23.4 | 8.7 | 13.2 |
| 6.6 | 33.1 | 7.7 | 22.7 | 8.8 | 11.7 |
| 6.7 | 31.7 | 7.8 | 22.1 | 8.9 | 9.7 |
| 6.8 | 30.4 | 7.9 | 21.5 | 9.0 | 7.7 |
| 6.9 | 29.6 | 8.0 | 9.1 | 4.7 | |
| 7.0 | 28.1 | 8.1 | 20.2 | 9.2 | 1.8 |
*Для получения раствора индикатора с заданным значением рН в мерную колбу на 50 мл налить 5 мл раствора индикатора, необходимое количество раствора КН2PO4 и довести до метки раствором буры.
Контрольные вопросы к теме: "Определение термодинамической константы кислотной диссоциации одноцветного индикатора"
1. Что такое кислоты и основания с точки зрения теории Бренстеда-Лоури?
2. Чем характеризуется сила кислоты и основания? Физический смысл величин 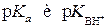 .
.
3. Кислотность водных растворов, величина рН среды. Буферные растворы, зависимость рН и величины буферной емкости от их состава.
4. Фотоколориметрический метод анализа, закон светопоглощения.
5. Индикаторы кислотности среды. Фотоколориметрическое определение термодинамической константы кислотности одноцветного индикатора.
6. Ход работы и обработка полученных данных.
Литература
1. Практические работы по физической химии /Под ред. К.П.Мищенко, А.А.Равделя, А.М.Пономаревой. -Л.: Химия, 1982.-С. 153-157, 173-179.
РАБОТА № 6
КАЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ТЕПЛОТЫ НЕЙТРАЛИЗАЦИИ
1. Теоретическая часть
1.1. Некоторые общие понятия.
Внутренняя энергия системы. Это суммарная энергия системы, складывающаяся из внутриядерной энергии, энергии электронных оболочек атомов, колебательной энергии атомов в молекулах, вращательной и поступательной энергии молекул. Она измеряется в единицах энергии [Дж] и равна общей энергии системы за вычетом кинетической и потенциальной ее энергии как единого целого. Для идеального газа внутренняя энергия зависит только от температуры и является функцией состояния. Элементарное изменение внутренней энергии это полный дифференциал (интегрирование от состояния 1 до состояния 2 дает конечное изменение внутренней энергии, а круговой интеграл равен нулю):
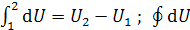 =
= 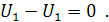
Для любой системы внутренняя энергия при температуре Т равна ее величине при абсолютном нуле 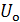 плюс ее приращение при нагревании системы до рассматриваемой температуры
плюс ее приращение при нагревании системы до рассматриваемой температуры 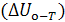 :
:
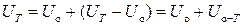 .
.
Абсолютная величина внутренней энергии системы неизвестна, поскольку неизвестной является энергия 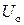 . В то же время ее изменение (Δ U) в ходе любого процесса легко измеримо (см. далее).
. В то же время ее изменение (Δ U) в ходе любого процесса легко измеримо (см. далее).
Теплота. Это энергия, передаваемая в виде теплоты термодинамической системе от среды (в этом случае она положительна) или среде от системы (отрицательна) в ходе какого-либо термодинамического процесса. Теплота является функцией процесса. Ее элементарное приращение не является полным дифференциалом и обозначается значком 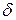 (интегрирование от состояния 1 до состояния 2 дает величину теплоты процесса, а круговой интеграл может быть и не равен нулю):
(интегрирование от состояния 1 до состояния 2 дает величину теплоты процесса, а круговой интеграл может быть и не равен нулю):

Работа. Это энергия, передаваемая в виде механической работы термодинамической системе от среды (в этом случае она отрицательна) или от системы к среде (положительна). Работа также является функцией процесса, как и теплота (см. выше):
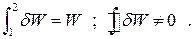
И теплота и работа измеряются в единицах энергии - джоулях [Дж].
1.2. Первое начало термодинамики.
Первое начало термодинамики является следствием универсального закона сохранения энергии и одна из его формулировок гласит: теплота, подведенная к системе, расходуется на изменение ее внутренней энергии (в простейшем случае - на ее нагревание) и на совершение системой работы.
Математическое выражение первого начала термодинамики в соответствии с этой формулировкой для элементарного изменения в системе имеет следующий вид:
для конечного изменения, то есть для термодинамического процесса (после интегрирования этого уравнения от состояния 1 до состояния 2):
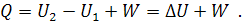
В простейшем случае, когда работа, совершаемая системой, состоит только из работы расширения, то есть когда 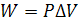 , математическое выражение первого начала термодинамики приобретает вид:
, математическое выражение первого начала термодинамики приобретает вид:
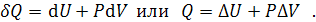
В дальнейшем будем рассматривать только этот простейший случай.
Для изобарного процесса, когда 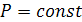 , математическое выражение первого начала термодинамики приобретает вид:
, математическое выражение первого начала термодинамики приобретает вид:
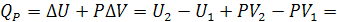
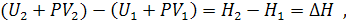
где 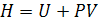 - энтальпия системы, равная сумме внутренней ее энергии
- энтальпия системы, равная сумме внутренней ее энергии 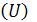 и потенциальной энергии, обусловленной энергией сжатого газа
и потенциальной энергии, обусловленной энергией сжатого газа 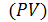 . Как и внутренняя энергия, энтальпия измеряется в [Дж]. Абсолютное значение энтальпии не может быть определено по той же причине, что и для внутренней энергии. В случае практически несжимаемых жидких и твердых веществ H приблизительно равна U. Энтальпия является функцией состояния, как и U, P, V.
. Как и внутренняя энергия, энтальпия измеряется в [Дж]. Абсолютное значение энтальпии не может быть определено по той же причине, что и для внутренней энергии. В случае практически несжимаемых жидких и твердых веществ H приблизительно равна U. Энтальпия является функцией состояния, как и U, P, V.
Таким образом, теплота изобарного процесса равна изменению энтальпии системы и наоборот - изменение энтальпии системы в изобарном процессе в точности равно теплоте процесса:
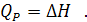
Для изохорного процесса, когда V = constиd V = 0, математическое выражение первого начала термодинамики приобретает вид:
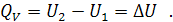
Теплота изохорного процесса равна изменению внутренней энергии системы или проще - в изохорном процессе, когда работа расширения системы равна нулю, теплота подведенная к системе расходуется только на ее нагревание.
Для изобарно-изотермического процесса, протекающего при данной температуре и данном давлении (T = const, P = const), изменение объема системы происходит только при изменении числа молей газа (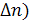 в ходе химиической реакции или физического процесса. Для идеальных газов
в ходе химиической реакции или физического процесса. Для идеальных газов 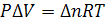 . Следовательно, первое начало термодинамики в этом случае может быть записано в форме
. Следовательно, первое начало термодинамики в этом случае может быть записано в форме
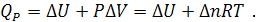
Поскольку 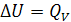 , соотношение между тепловыми эффектами процессов, протекающих при постоянном давлении и постоянном объеме, следующее:
, соотношение между тепловыми эффектами процессов, протекающих при постоянном давлении и постоянном объеме, следующее:
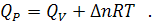
1.3. Теплоемкость, зависимость ее от температуры, изобарная, изохорная теплоемкости, соотношение между ними.
Теплоемкостью системы называется количество теплоты, необходимое для нагревания ее на 1 градус. Теплоемкость обозначается буквой С и измеряется в [Дж/град].
Удельной теплоемкостью вещества называется количество теплоты, необходимое для нагревания единицы количества вещества на 1 градус.
В зависимости от единицы измерения количества вещества удельная теплоемкость измеряется в [Дж/кг×град], [Дж/см3×град], [Дж/моль×град] и т.д. и обозначает количество тепла, необходимое для нагревания на 1 градус одного кг, одного см3 или одного моля вещества. В последнем случае теплоемкость называется мольной.
Удельная теплоемкость зависит от физико-химических свойств вещества (разные вещества имеют разную теплоемкость), от температуры (теплоемкость с температурой растет), от агрегатного состояния вещества (жидкая и газообразная формы вещества имеют разную теплоемкость даже при одной и той же температуре), а для газов и от способа нагревания (при постоянном давлении или при постоянном объеме).
Различают истинную и среднюю теплоемкость. Истинная теплоемкость определяется выражением
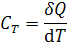
и представляет собой теплоемкость при данной температуре. Средней мольной теплоемкостью в интервале температур от T 1 до T 2 называют отношение количества тепла, необходимого для нагревания 1 моля вещества от температуры T 1 до T 2 , к разности температур:
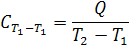 .
.
Если нагревание проводится при постоянном давлении, теплоемкость называется изобарной:
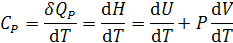 .
.
Соответственно, изохорная теплоемкость:
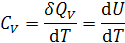 .
.
Из сопоставления выражений для 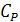 и
и 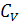 следует:
следует:
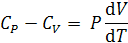 .
.
Для жидкостей и твердых веществ изменение объема 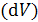 при нагревании 1 моля вещества на 1 градус очень мало, поэтому с точностью до нескольких процентов величины CP и CV можно считать равными друг другу:
при нагревании 1 моля вещества на 1 градус очень мало, поэтому с точностью до нескольких процентов величины CP и CV можно считать равными друг другу: 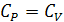 . Для газов изменением объема при нагревании пренебречь нельзя и разница между изобарной и изохорной теплоемкостью оказывается достаточно большой. Если считать газ идеальным, то для одного моля выполняется соотношение PV = RT. Отсюда
. Для газов изменением объема при нагревании пренебречь нельзя и разница между изобарной и изохорной теплоемкостью оказывается достаточно большой. Если считать газ идеальным, то для одного моля выполняется соотношение PV = RT. Отсюда
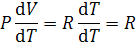 .
.
Следовательно, для газов 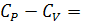 8.31 Дж/моль×град. Изобарная теплоемкость больше изохорной на величину R, то есть на величину работы расширения 1 моля идеального газа при нагревании его на 1 градус.
8.31 Дж/моль×град. Изобарная теплоемкость больше изохорной на величину R, то есть на величину работы расширения 1 моля идеального газа при нагревании его на 1 градус.
Зависимость теплоемкости от температуры выражается эмпирическими уравнениями:
 и
и
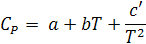 .
.
соответственно для органических и неорганических веществ. Здесь a, b, c и с' - константы, приведенные в справочнике.
1.4. Теплота растворения, разведения, нейтрализации.
Мольная теплота растворения вещества - это теплота, которая выделяется или поглощается при растворении 1 моля вещества в каком-либо растворителе (чаще всего в воде). Растворение электролитов (солей, кислот, оснований) в воде сопровождается тепловым эффектом вследствие того, что при этом происходит разрушение кристаллической решетки (для твердых веществ) или диссоциация молекул (для жидкостей), на что затрачивается энергия. Теплота при этом поглощается и D H (дисс.)> 0. Гидратация образующихся ионов приводит к выделению тепла (D H (гидр.)< 0). Например:
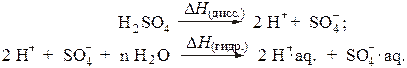
Суммарный тепловой эффект является суммой этих теплот и зависит от соотношения их величин и может быть как положительной, так и отрицательной:
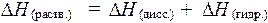
Величина теплового эффекта растворения одного моля вещества зависит от его физико-химических свойств (серная кислота, например, растворяется с выделением тепла, а тиосульфат натрия - с поглощением) и от концентрации образующегося раствора. Чем разбавленнее образующийся раствор, тем больше тепловой эффект растворения и наоборот. В справочниках приводятся стандартные (для 298 К) значения мольных интегральных теплот растворения 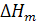 [Дж/моль] для различных веществ и различных моляльных концентраций (m) образующихся растворов. Для вычисления тепловых эффектов растворения определяют число молей вещества (n, молей) в заданном его количестве и моляльную концентрацию образующегося раствора (m, молей/кг воды). По величине m в справочнике находят интегральную мольную теплоту растворения . Теплоту растворения данного количества вещества в заданном количестве воды находят умножением мольной теплоты растворения на количество молей вещества:
[Дж/моль] для различных веществ и различных моляльных концентраций (m) образующихся растворов. Для вычисления тепловых эффектов растворения определяют число молей вещества (n, молей) в заданном его количестве и моляльную концентрацию образующегося раствора (m, молей/кг воды). По величине m в справочнике находят интегральную мольную теплоту растворения . Теплоту растворения данного количества вещества в заданном количестве воды находят умножением мольной теплоты растворения на количество молей вещества:
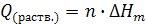 .
.
Мольная теплота разведения раствора вещества водой от концентрации m 1 до концентрации m 2 может быть найдена из теплот растворения. Представим, что один моль вещества растворяют в воде с образованием разбавленного раствора с концентрацией m 2 двумя путями - непосредственно добавлением воды (путь 1), а также с предварительным получением более концентрированного раствора с концентрацией m 1 и последующим его разбавлением до концентрации m 2 (путь 2):
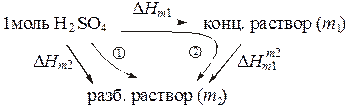
Из закона Гесса следует
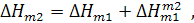
откуда

При разбавлении раствора, содержащего n молей вещества, выделяется или поглощается в n раз больше тепла:
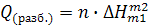 .
.
Теплота нейтрализации кислоты -это теплота реакции взаимодействия одного ее грамм-эквивалента с основанием. Сильные кислоты и сильные основания нацело диссоциируют в воде с образованием соответствующих гидратированных ионов, поэтому теплота нейтрализации сильной кислоты - это теплота образования одного моля воды из гидратированных гидроксид- и гидроксоний-ионов:
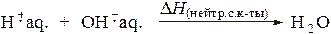
По этой причине вне зависимости от природы сильной кислоты и сильного основания теплота нейтрализации одного ее грамм-эквивалента составляет 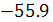 кДж/г-экв.
кДж/г-экв.
В случае слабой кислоты нейтрализация включает 2 стадии - диссоциацию кислоты с образованием гидратированных ионов и последующее взаимодействие катиона гидроксония с гидроксид-анионом. Например, нейтрализация синильной кислоты:
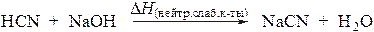
протекает следующим образом:
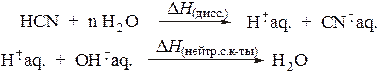
В соответствии с этим теплота нейтрализации слабой кислоты включает в себя теплоту диссоциации:
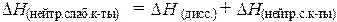
и поэтому может быть больше или меньше чем 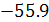 кДж/г-экв.
кДж/г-экв.
2. Определение теплот процессов
Теплоты самых различных процессов могут быть определены на опыте при помощи установки, называемой калориметром. Установка представляет собой сосуд, помещенный в теплоизолирующую оболочку. В простейшем случае сосуд это стакан, снабженный мешалкой и термометром. В результате протекания процесса и, следовательно, выделения или поглощения тепла, температура в сосуде изменяется. Если пренебречь изменением теплоемкости в результате этого, как правило, небольшого изменения температуры, можно записать:
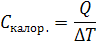 ,
,
где 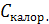 - теплоемкость калориметра. В зависимости от того в открытом или закрытом сосуде проводится процесс, это должна быть либо , либо , соответственно (для процесса, проводимого в жидкой фазе разницей между и можно пренебречь);
- теплоемкость калориметра. В зависимости от того в открытом или закрытом сосуде проводится процесс, это должна быть либо , либо , соответственно (для процесса, проводимого в жидкой фазе разницей между и можно пренебречь);  - тепловой эффект процесса (
- тепловой эффект процесса (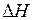 или
или 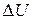 соответственно);
соответственно); 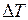 - изменение температуры в ходе протекания процесса. Если процесс проводится в открытом сосуде при постоянном давлении, тогда тепловой эффект процесса расходуется на изменение энтальпии и равен произведению теплоемкости калориметра на изменение его температуры:
- изменение температуры в ходе протекания процесса. Если процесс проводится в открытом сосуде при постоянном давлении, тогда тепловой эффект процесса расходуется на изменение энтальпии и равен произведению теплоемкости калориметра на изменение его температуры:
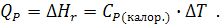
Таким образом, определение теплового эффекта сводится к определению теплоемкости калориметрической установки и величины изменения ее температуры в ходе протекания изучаемого процесса (
| <== предыдущая | | | следующая ==> |
| образования ΔG°298 некоторых веществ | | | УДК 544 (076) |
Date: 2015-09-18; view: 1788; Нарушение авторских прав