Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Вывод: При реакции поликонденсации образуются побочные продукты, а при реакции полимеризации - нет
|
|
Существует система классификации реакций полимеризации на реакции цепной полимеризации и реакции ступенчатой полимеризации.
Различие между реакциями цепной полимеризации и ступенчатой полимеризации несколько более запутано, чем между реакциями полимеризации и поликонденсации. Это различие заключается примерно в следующем:
При цепной полимеризации мономеры становятся частью молекулы полимера постепенно, одна за другой, так что в данный момент времени к молекуле полимера присоединяется только одна молекула мономера.
Пример картины цепной полимеризации, анионной полимеризации стирола с образованием полистирола: 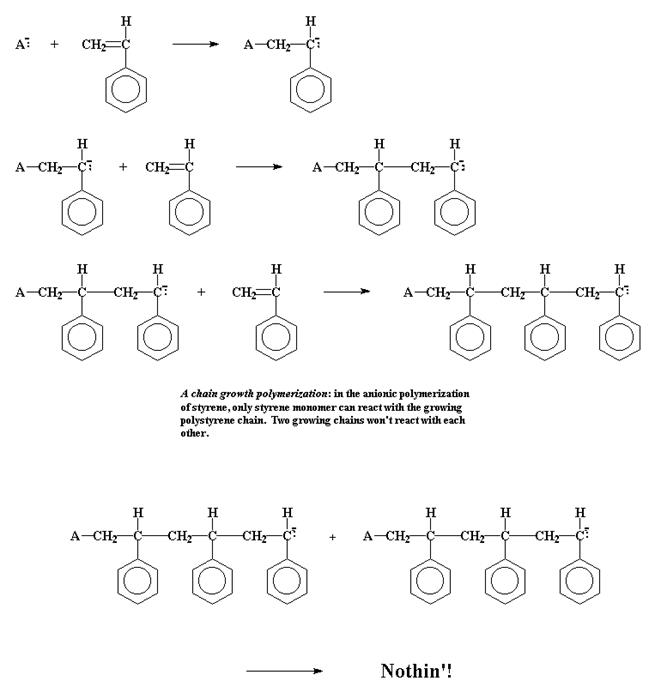
Но при ступенчатой полимеризации все происходит несколько более сложным образом. Посмотрим на ступенчатую полимеризацию двух мономеров, хлорангидрида терефталевой кислоты и этиленгликоля, с образованием полиэфира под названием полиэтилентерефталат. Первое, что происходит при этой реакции, это взаимодействие двух мономеров с образованием димеров. Звучит достаточно просто.
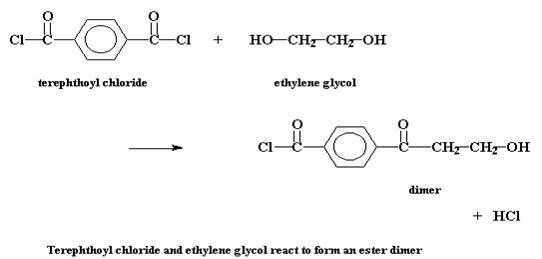
Будь эта система такой, в которой происходит цепная полимеризация, на этой стадии реакции могло бы произойти только одно: третья молекула мономера присоединилась бы к димеру, чтобы образовать тример, затем четвертая - чтобы образовать тетрамер и так далее. Но в свободной стране ступенчатой полимеризации с этим димером может произойти много разных процессов. Конечно же, он может прореагировать с одним из мономеров, чтобы образовать таким образом тример:
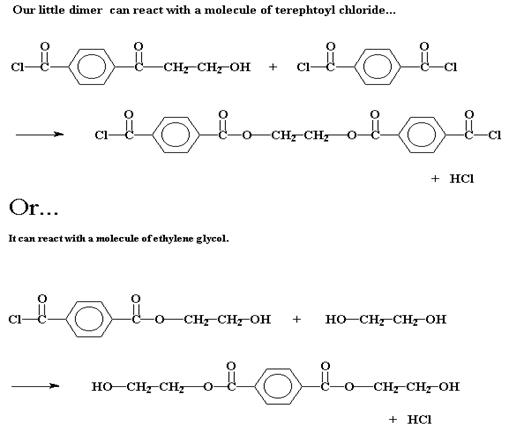
Но он может также поступить иначе. Он может провзаимодействовать с другим димером, чтобы образовать тетрамер:
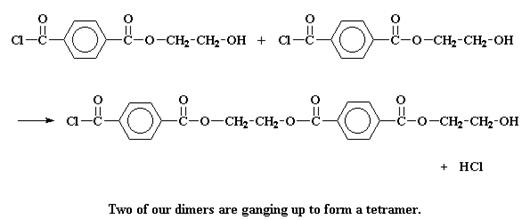
Или он может поступить совсем сумасшедшим образом и вступить в реакцию с тримером с образованием пентамера.
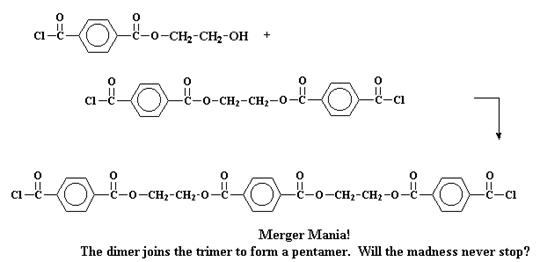
Делая процесс все более и более запутанным, эти тетрамеры и пентамеры могут провзаимодействовать между собой и образовать олигомеры еще больших размеров. И, таким образом, они продолжают расти и расти, пока олигомеры не становятся достаточно большими, чтобы иметь уже право называться полимерами.
Вы просто подумайте о том, как банки постоянно сливаются с другими банками и становятся при этом все крупнее и крупнее. Это поможет вам лучше представить процесс ступенчатой полимеризации.
Вывод: Основное отличие заключается в том, что при ступенчатой полимеризации растущие молекулярные цепи могут взаимодействовать между собой, образуя еще более длинные цепи. И это справедливо для цепей любой длины. Мономер или димер может вступить в реакцию точно также, как и цепочка, состоящая из сотен мономерных звеньев. Но при цепной полимеризации только мономеры могут взаимодействовать с растущей цепочкой макромолекулы. Сами растущие цепочки не могут взаимодействовать между собой так, как это они могут делать при ступенчатой полимеризации.
Непримиримые противоречия:
Ступенчатая полимеризация для получения полиэфира привела к образованию побочного продукта, газообразного хлористого водорода HCl. Вследствие этого, конечно же, эту реакцию ступенчатой полимеризации можно отнести к типу реакции поликонденсации.
Вы также могли заметить, что наша реакция цепной полимеризации не привела к образованию побочного продукта. Да, конечно, эта ступенчатая реакция полимеризации является также реакцией полимеризации (а не поликонденсации), то есть реакцией присоединения.
Довольно легко из этого сделать вывод о том, что ступенчатая полимеризация и поликонденсация - это одно и то же, а также о том, что полимеризация присоединения и цепная полимеризация - тоже ничем друг от друга не отличаются. Однако это будет как раз неверно. Ведь существуют реакции полимеризации присоеднинения, которые являются ступенчатыми реакциями полимеризации. Одним из примеров может служить полимеризация, в результате которой образуются полиуретаны. Кроме того, существуют также реакции поликонденсации, которые являются цепными реакциями полимеризации. Попытки как-то примирить между собой систему классификации на реакции цепной и ступенчатой полимеризации и систему классификации на реакции полимеризации и поликонденсации - это пустая трата времени. У каждой из этих систем свои критерии, и различия, характерные для одной системы, не всегда будут тождественны различиям, характерным для другой системы классификации.
Поэтому и не думайте связать эти две системы. Просто надо знать, что реакции образования полимеров могут быть либо цепными, либо ступенчатыми, с одной стороны, а с другой стороны, они могут быть либо реакциями полимеризации, либо поликонденсации.
№3. Побочные реакции, осложняющие поликонденсацию.
В общем виде схема основной реакции П.-роста цепи-м. б. представлена след, образом.:
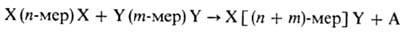
(n и m-любое целое число, включая единицу, X и Y-исходные функц. группы, А-низкомол. продукт поликонденсации.). При этом взаимодействии мономеров друг с другом или с образовавшимися олигомерами и последних между собой подчиняется практически одним и тем же законам.
Поскольку при поликонденсации мономеры исчерпываются уже при невысоких степенях завершенности реакции, рост цепи высокомолекулярного полимера происходит преимущественно в результате многократного соединения между собой олигомерных или полимерных молекул по концевым функц. группам (принцип многократного удвоения), при этом число молекул в системе уменьшается (в этом ступенчатый характер поликонденсации.). Уменьшается в ходе поликонденсации и количество исходных функц. групп-реакционных (активных) центров, хотя в ряде случаев образующиеся при поликонденсации связи реагируют как между собой, так и с исходными реакц. центрами. Росту полимерной цепи при равновесной поликонденсации сопутствует обратная реакция полимера с выделяющимся низкомол. продуктом, что ограничивает мол. массу полимера.
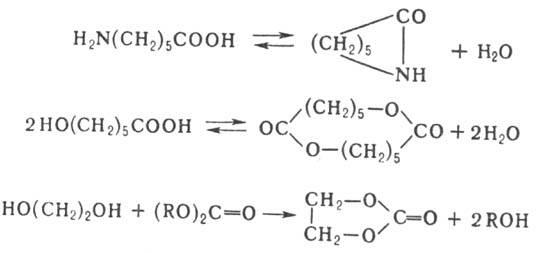
При поликонденсации функц. группы мономеров, олигомеров и полимерных цепей расходуются не только на рост цепи, но и на побочные реакции (реакции с примесями или со специально введенными в процесс веществами, декарбоксилирование карбоновых кислот и др.), что также лимитирует мол. массу образующегося полимера. При поликонденсации возможны также циклизация и обменные реакции. Циклизация м. б. внутримолекулярной, когда кольца образуются при р-ции функц. групп одной молекулы, или межмолекулярной при взаимод. двух или более молекул одинаковой или разл. природы, напр.:
Возможность циклизации определяется соотношением двух факторов:
1) снижения вероятности образования цикла по мере увеличения его размера (увеличение энтропии активации);
2) напряженности цикла, к-рая уменьшается при увеличении его размера вплоть до 6-членного, затем возрастает при изменении числа членов в цикле от 6 до 9-11, а затем вновь снижается при переходе к еще большим циклам. В результате совместного действия обоих этих факторов при получении методом поликонденсации различных полимеров возможно образование больших циклов (20-40-членных). Циклообразованию способствует проведение реакции в сильно разб. р-рах (см. Краун-эфиры).
Обменные реакции особенно эффективны при повышенных температурах поликонденсации. Их делят на два основных типа:
1) реакции обмена образовавшихся при поликонденсации групп (сложноэфирной, амидной или др.) и даже некоторых циклов (напр., имидного) с функц. группами мономеров или примесей (напр., алкоголиз, ацидолиз, аминолиз);
2) реакции межцепного обмена между образовавшимися при поликонденсации одно- или разнотипными группами (напр., эфиролиз, амидолиз). Эффективность обменных реакций зависит от соотношения скоростей основной и побочных реакций. Обменные реакции могут существенно влиять на мол. массу и MMP поликонденсац. полимера, микроструктуру сополимера. В ряде случаев обменные реакции положены в основу получения поликонденсац. гомо- и сополимеров, напр, синтез поли-этилентерефталата переэтерификацией диметилтерефталата этиленгликолем.
Ограничение роста полимерной цепи м. б. обусловлено и чисто физическими причинами, напр. преждевременным выпадением полимера из реакционной среды в осадок при поликонденсации в растворе (особенно если это сопровождается его кристаллизацией), однако если выпадающий из раствора полимер набухает в реакц. среде, рост цепи часто не прекращается.
№4. Механизмы реакций поликонденсации в кислой и щелочной среде фенола и формальдегида.
Фенолформальдегидные смолы - продукты поликонденсации фенола с формальдегидом. Реакция проводится в присутствии кислых (соляная, серная, щавелевая и другие кислоты) или щелочных катализаторов (аммиак, гидроксид натрия, гидроксид бария). При избытке фенола и кислом катализаторе образуется линейный полимер – новолак, цепь которого содержит приблизительно 10 фенольных остатков, соединенных между собой метиленовыми мостиками:

Новолаки – термопластичные полимеры, которые сами по себе не способны переходить в неплавкое и нерастворимое состояние. Но они могут превращаться в трехмерный полимер при нагревании их с дополнительной порцией формальдегида в щелочной среде.
При использовании щелочных катализаторов и избытка альдегида в начальной стадии поликонденсации получаются линейные цепи резола, которые при дополнительном нагревании "сшиваются" между собой за счет групп CH2OH, находящихся в пара-положении фенольного кольца, с образованием трехмерного полимера – резита:
Таким образом, резолы являются термореактивными полимерами.
Фенолоформальдегидные полимеры применяются в виде прессовочных композиций с различными наполнителями, а также в производстве лаков и клея.
Свойства: Отвержденные смолы характеризуются высокими тепло-, водо- и кислостойкостью, а в сочетании с наполнителями и высокой механической прочностью.
Применение: Из фенолформальдегидного полимера, добавляя различные наполнители, получают фенолформальдегидные пластмассы, т. н. фенопласты. Их применение очень широко. Это: шарикоподшипники, шестерни и тормозные накладки для машин; хороший электроизоляционный материал в радио- и электротехнике. Изготовляют детали больших размеров, телефонные аппараты, электрические контактные платы. Для склеивания пенополистирольных плит, применяемых для изготовления моделей в литейном производстве.
Получение фенолформальдегидной смолы:
1. В пробирку помещают 10 капель жидкого фенола и 8 капель 40% формальдегида. Смесь нагревают на водяной бане до растворения фенола. Через 3 минуты в пробирку добавляют 5 капель концентрированной соляной кислоты и помещают ее в стакан с холодной водой. После образования в сосуде двух четких фаз следует слить воду и вылить полимер из пробирки. В течение нескольких минут образовавшаяся новолачная смола затвердевает.
2. В небольшую колбочку помещают 15 г фенола и 25 мл концентрированного раствора формалина и нагревают (под тягой) на горелке, периодически встряхивая содержимое колбы. Добавляют 1-2 мл соляной кислоты и продолжают нагревание. Вначале реакция идет бурно и смесь в колбе становится однородной. Через некоторое время (около 10 минут) на дне колбы образуется смолистый осадок. Верхний слой жидкости сливают и быстро извлекают смолу, которая на воздухе густеет и постепенно затвердевает.
Фенолформальдегидные смолы [-C6H3(OH)-CH2-]n – продукты поликонденсации фенола C6H5OH с формальдегидом CH2=O.
Взаимодействие фенола с формальдегидом идет по схеме:
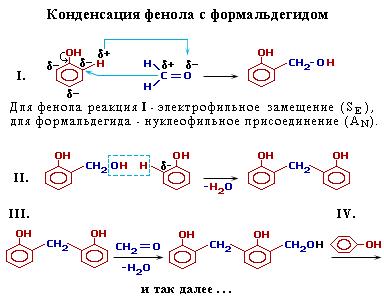
Роль реакционноспособных функциональных групп в этих соединениях играют:
в феноле – три С-Н-связи в орто- и пара-положениях (легче идет замещение в двух орто-положениях);
в формальдегиде – двойная связь С=О, способная к присоединению по атомам С и О.
Это определяет возможность образования цепных макромолекул по схеме поликонденсации: (нет схемы)
Реакция проводится в присутствии кислых (соляная, серная, щавелевая и другие кислоты) или щелочных катализаторов (аммиак, гидроксид натрия, гидроксид бария).
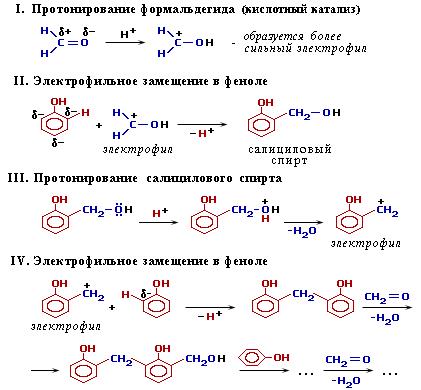
См. механизм конденсации в условиях кислотного катализа.
При избытке фенола и кислом катализаторе образуется линейный полимер – новолак, цепь которого содержит приблизительно 10 фенольных остатков, соединенных между собой метиленовыми (-СН2-) мостиками.
Новолаки – термопластичные полимеры, которые сами по себе не способны переходить в неплавкое и нерастворимое состояние. Но они могут превращаться в трехмерный полимер при нагревании их с дополнительной порцией формальдегида в щелочной среде. При использовании щелочных катализаторов и избытка альдегида в начальной стадии поликонденсации получаются линейные цепи резола: (нет схемы)
При дополнительном нагревании эти цепи "сшиваются" между собой за счет групп -CH2OH, находящихся в пара-положении фенольного кольца, с образованием трехмерного полимера – резита: (нет схемы)
Таким образом, резолы являются термореактивными полимерами. Полимеры, которые при повышенной температуре приобретают пространственную (сетчатую) структуру и становятся неплавкими и нерастворимыми, называются термореактивными.
Фенолоформальдегидные полимеры применяются в виде прессовочных композиций с различными наполнителями, а также в производстве лаков и клея.
Механизм конденсации фенола с формальдегидом в условиях кислотного катализа:
№5. Химизм поликонденсации фталевого ангидрида и глицерина, адипиновой кислоты и гексаметилендиамина, мочевины и формальдегида, терефталевой кислоты и этиленгликоля.
Фталевый ангидрит+глицерин=алкидные смолы
Алкидные смолы, продукты взаимодействия многоосновных карбоновых кислот, многоатомных спиртов (полиолов) и одноосновных высших жирных кислот. Многоосновные кислоты используют для синтеза алкидных смол обычно в виде ангидридов, высшие жирные кислоты - в виде индивидуальных соединений или в составе растит, масел (полных сложных эфиров этих кислот и глицерина-триглицеридов). Наиболее распространенные алкидные смолы получают из фталевого ангидрида и глицерина (глифталевые смолы), и пентаэритрита (пентафталевые смолы) или триметилолпропана, называемого также этриолом (этрифталевые смолы). Алкидные смолы, в состав которых входят остатки кислот высыхающих или полувысыхающих масел (например, льняного, тунгового, подсолнечного), называют высыхающими; алкидные смолы, содержащие остатки кислот невысыхающих масел (напр., касторового), относят к невысыхающим. По количеству остатков кислот (жирности) алкидные смолы подразделяют на сверхтощие (< 35%), тощие (35-45%), средние (46-55%), жирные (56-70%) и очень жирные (> 70%).
Алкидные смолы - высоковязкие липкие продукты от светло-желтого до коричневого цвета; мол. м. 1500-5000. Тощие смолы растворяются лишь в ароматических углеводородах (толуоле, ксилоле. сольвенте), жирные-в алифатических (например, уайт-спирите), смолы средней жирности-в смесях ароматических и алифатических углеводородов.
Применяют алкидные смолы в качестве пленкообразователей лакокрасочных материалов. Высыхающие смолы образуют покрытия в результате окислительно-полимеризационных процессов с участием ненасыщенных связей жирных кислот, невысыхающие - в результате улетучивания растворителя или(и) поликонденсации, в которой участвуют функциональные группы смолы и введенного в материал отвердителя.
Получение. Осн. реакция при получении алкидные смолы-конденсационных теломеризация (полиэтерификация), в которой высшая жирная кислота служит телогеном, например:
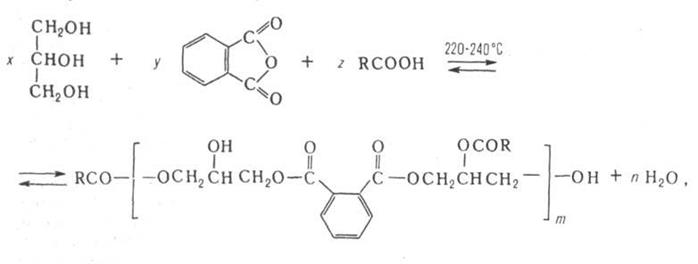 где R-алкил или алкенил, m 2.
где R-алкил или алкенил, m 2.
В промышленности применяют два метода синтеза - жирнокислотный и алкоголизный. В первом случае исходят из свободной жирной кислоты, во втором из раститительного масла, которое сначала подвергают алкоголизу полиолом, а затем образовавшиеся неполные эфиры вводят в реакцию с фталевым ангидридом. При синтезе алкидные смолы на основе касторового масла (так называемых резиловых смол) алкоголиз масла не требуется, так как в полиэтерификацию с ангидридом вступают группы ОН остатков рицинолевой кислоты.
Катализаторы алкоголиза-РbО, CaO, Na2CO3 и другие, полиэтерификации - кислоты-реагенты; иногда дополнительно используют также минеральные кислоты или их соли и др. Реакция ускоряется и в присутствии 1-2% малеинового ангидрида, который образует аддукты Дильса - Альдера с жирными кислотами, содержащими сопряженные двойные связи. или присоединяется по метиленовым группам кислот с изолированными двойными связями. В последнем случае образуются трехосновные алкенилянтарные кислоты, повышающие среднюю функциональность системы, что ускоряет рост вязкости реакционной массы. Кроме того, малеиновый ангидрид, взаимодействуя с хромофорными группами масел и разрушая систему сопряжения в них, способствует получению более светлых алкидные смолы.
Алкидные смолы синтезируют в условиях, обеспечивающих получение продукта с кислотным числом в пределах 10-20, т.к. из-за присутствия в смоле большого числа свободных групп СООН может повышаться вязкость при хранении лакокрасочных материалов, содержащих пигменты основного характера. Образование смолы с низким кислотным числом достигается введением в исходную реакционную смесь избытка полиола.
Синтезируют алкидные смолы в основном по периодической схеме в реакторах с высокотемпературным обогревом. Распространены также полунепрерывные схемы с применением реакторов большой единичной мощности (32 м3 и более). Фталевый ангидрид применяют в виде гранул или расплава. в последнем случае может быть автоматизирована его загрузка и снижена общая продолжительность процесса.
В производстве алкидные смолы жирнокислотным методом все компоненты загружают одновременно; температура полиэтерификации 210-250°С. Достоинства метода: одностадийность; возможность получения пентафталевых смол. не содержащих остатков глицерина; однородность алкидные смолы и их светлая окраска, обусловленные сравнительно невысокими температурами синтеза; высокая стабильность процесса. Недостаток: необходимость предварительного расщепления раститительных масел, что сопровождается их потерями и требует применения специальной коррозионностойкой аппаратуры. В отечественной промышленности значительно шире применяют алкоголизный метод. Триглицерид реагирует с полиолом в расплаве при 240-260°С до образования продукта, растворимого в этаноле. Недостатки метода: двустадийность; плохая воспроизводимость, связанная с побочными реакциями при алкоголизе и с потерями полиола на этой стадии; присутствие в пента- и глифталевых смолах остатков глицерина.
Для удаления воды, образующейся при полиэтерификации, используют блочный или азеотропный способ. В первом случае в реакторе создают небольшой вакуум и барботируют через реакционную массу N2 или СО2, во втором - вводят в реактор ксилол (2-3% от реакционной массы), образующий с водой азеотропную смесь, которую отгоняют, охлаждают и разделяют: ксилол с растворенными в нем органическими веществами возвращают в реактор, а загрязненную воду удаляют. Преимущества азеотропного способа: меньшее количество сточных вод, значительно меньшие потери фталевого ангидрида, возможность получения более светлых алкидных смол. Однако повышенная пожароопасность, более дорогое аппаратурное оформление и трудность регулирования работы азеотропной системы обусловливают широкое использование блочного способа. Растворы алкидных смол (алкидные лаки) получают в смесителях, куда смола поступает из реактора самотеком под слой предварительно налитого растворителя. Лаки центрифугируют, фильтруют и иногда подвергают отстаиванию.
Способы модифицирования. Для этой цели применяют: 1) введение различных сомономеров при синтезе смолы; 2) взаимодействие готовых смол с модифицирующими веществами; 3) смешение смол с другими пленкообразователями (см. табл.). На стадии синтеза смол до 30% жирных кислот заменяют бензойной кислотой, кислотами канифоли, нафтеновыми кислотами и другими или вместо части фталевого ангидрида применяют диизоцианаты либо алкоксисилоксаны (синтез соотв. алкидно-уретановых, или уралкидных, и алкидно-силоксановых смол с участием групп ОН продукта алкоголиза или образующейся смолы). Готовые алкидные смолы (главным образом на основе тунгового или дегидратированного касторового масла. сополимеризуют по двойным связям жирных кислот с метакрилатами или стиролом в растворе в присутствии радикальных инициаторов. Возможна также прививка стирола по двойным связям введенного в состав смолы малеинового ангидрида.
Адипиновая кислота+гексаметилендиамин=полигексаметиленадипинамид
ПОЛИГЕКСАМЕТИЛЕНАДИПИНАМИД (полигексаме-тиленадипамид, полиамид-6,6, найлон-6,6, зайтел 101, анид и др.) [— HN(CH2)6NHCO(CH2)4CO— ]n, аморфно-кристал-лич. бесцв. роговидный полимер. мол.м. (15-30)·103; т.стекл. 45-700C, т.пл. 2650C; плотн. 1,130-1,150 г/см3; раств. в феноле. м-крезоле, муравьиной кислоте, конц. H2SO4. Равновесное влагосодержание 3,5-4% при 200C и 65%-ной относит. влажности воздуха. полигексаметиленадипинамид сочетает высокую мех. прочность, в т.ч. при ударных нагрузках, эластичность и износостойкость, что обусловливает хорошие эксплуатац. свойства изделий из него. Для полигексаметиленадипинамид характерны: 75-110 МПа, 100-120 МПа, 100-110 МПа; модуль упругости 1500-1600 МПа; ударная вязкость 100-160 кДж/м2; относит. удлинение 80-100%; твердость по Бринеллю 80-100 МПа; теплостойкость по Вика 2300C, по Мартенсу 650C; коэф. теплопроводности 0,24-0,29 Вт/(м·К); 0,08 кДж/(кг·К); температурный коэф. линейного расширения (5 - 6)· 10 -5 оC -1; 1013 - 1014 Ом·см; 0,02 при 1 МГц; e 4,0 при 1 МГц; электрич. прочность 20-25 кВ/мм; водопоглощение 7-8%; бензостойкость 0,4%. При нагр., под действием O2 воздуха. УФ и радиац. излучений полигексаметиленадипинамид подвергается деструкции, в результате чего снижается мол. масса и ухудшаются мех. свойства. Поэтому для увеличения срока службы изделий из полигексаметиленадипинамид в промышленности в него вводят разл. термо- и светостабилизаторы (ароматич. амины. фенолы и некоторые неорг. соли). Для придания повыш. гигроскопичности замещают водород амидной группы в полигексаметиленадипинамид на гидроксиметильную группу или полиэтиленоксидные цепи (р-цией с формальдегидом или этиленоксидом соотв.).
Осн. метод получения полигексаметиленадипинамид-равновесная гидролитич. поликонденсация гексаметилендиамина (Г) с адипиновой кислотой (А) в расплаве. Мономеры используют в виде соли (соль АГ с т.пл. 183-1840C), т.к. при этом точно соблюдается их эквимол. соотношение, что обеспечивает получение полигексаметиленадипинамид высокой мол. массы. Из полигексаметиленадипинамид формованием из расплава (см. Полиамидные волокна)производят волокна и: нити (на это тратят ~90% выпускаемого в мире полигексаметиленадипинамид), литьем под давлением и экструзией - шестерни, вкладыши подшипников, втулки, ленты, листы, трубы и др. изделия для машино- и приборостроения, электротехники.
Мочевина+формальдегид= мочевино-формальдегидные смолы.
МОЧЕВИНО-ФОРМАЛЬДЕГИДНЫЕ СМOЛЫ (карба-мидо-формальдегидные смолы, карбамидные смолы), син-тетич. термореактивные олигомерные продукты поликонденсации мочевины с формальдегидом. Образуются в результате поликонденсации первичных продуктов присоединения мочевины и формальдегида-метилмочевин H2NCONHCH2OH и СО(КНСН2ОН)2-друг с другом, мочевиной и формальдегидом. Состав, строение и свойства М.-ф.с. зависят от количеств. соотношения мочевины и формальдегида и условий синтеза (т-ра, продолжительность, концентрация исходных соед., рН реакц. среды). М.-ф.с.-смесь олигомеров разл. мол. массы линейной, разветвленной или циклоцепной структуры, содержащих реак-ционноспособные амино- и ОН-группы. В М.-ф. с. линейной и разветвленной структур остатки мочевины связаны мети-леновыми и метиленэфирными мостиками; в качестве структурных элементов содержат метилольные и гемиформаль-ные группы. В М.-ф.с. циклоцепной структуры, помимо указанных выше групп, имеются триазиноновые и уроновые циклы. В состав М.-ф.с. входят также своб. мочевина, метиленгликоль, а при избытке формальдегида-олигомерные полиоксиметиленгликоли.
Получают и применяют М.-ф.с. в виде водных преим. 40-70%-ных растворов (пожаро- и взрывобезопасны) и порошков. Технология произ-ва в значит. степени определяется назначением смолы. Так, М.-ф. с., используемые в качестве связующих и основы клеев, получают по непрерывной схеме след. образом: в 37%-ном формалине. рН которого водным раствором щелочи доводят до 5,0-6,0, растворяют мочевину, получая конденсац. раствор, имеющий рН 7,0-8,5 при молярном соотношении мочевина: формальдегид 1: (1,9-2,2). Процесс осуществляют в каскаде реакторов, куда непрерывно подают конденсац. раствор. На первой стадии процесса при 92-98 °С происходит постепенное снижение рН до 6,0. На второй стадии рН конденсац. раствора снижают до 4,5-5,0 с помощью водного раствора кислоты и завершают конденсацию после достижения заданных свойств смолы. Затем доводят рН до 7,0-8,0 водным раствором щелочи и при 80-90 °С и пониж. давлении осуществляют концентрирование реакц. массы, после чего вводят оставшуюся часть мочевины и проводят конденсацию при 60 °С. Полученную смолу охлаждают. При использовании на первой стадии аммиака вместо щелочи отпадает необходимость введения на второй стадии кислоты, т.к. требуемый диапазон рН 4,5-5,8 достигается самопроизвольно. Технология получения таких смол периодич. методом аналогична. М.-ф.с. для аминопластов получают растворением мочевины в 37%-ном формалине. рН которого водным раствором щелочи доводят до 6,6-7,0, с послед. конденсацией при 40-50°С и охлаждением полученного продукта. Порошкообразные М.-ф. с. получают сушкой (гл. обр. распылительной) их водных растворов в условиях, практически исключающих поликонденсацию.
Отверждаются М.-ф.с. при нагр. (120-140°C) или комнатной температуре в присутствии соединений преим. кислотного характера, например фосфорной, соляной, щавелевой, фталевой кислот, их солей (АlСl3, ZnCl2).
Получаемые в результате отверждения сетчатые полимеры бесцветны, светостойки, устойчивы в орг. растворителях и маслах, легко окрашиваются, однако имеют ряд недостатков-пониж. водостойкость, хрупкость, низкую устойчивость к деструктивным воздействиям, выделение своб. формальдегида и др. С целью устранения этих недостатков, а также придания требуемых свойств, например способности растворяться в орг. растворителях, увеличения гидрофобности и адге-зии, М.-ф. с. модифицируют либо при синтезе путем замены части мочевины на модифицирующий агент, либо уже готовый олигомер (напр., частичной этерификацией метилоль-ных групп). В зависимости от заданных свойств для модификации используют преим. одно- и многоатомные спирты (бутиловый, фурфуриловый, гликоли, глицерин), амины, амиды и др. производные карбоновых кислот, дициандиамид, меламин, гуанамины (см., например, Гуанамино-формальдегид-ные смолы), а также разл. высокомол. соединения.
Применяют М.-ф.с. в осн. как связующие в произ-ве древесностружечных плит и аминопластов, основу клеев для произ-ва фанеры и разл. деревянных конструкций (см., например, Древесина слоистая клееная, Древесные плиты, Древесные прессовочные массы). Смолы используют также в произ-ве декоративных бумажно-слоистых пластиков и синте-тич. шпона, влагостойкой бумаги, карбамидо-формальдегид-ных пенопластов, в текстильной промышленности для аппретирования тканей с целью придания им несминаемости. М.-ф.с., модифицированные бутиловым спиртом, используют для получения мочевино-алкидных лакокрасочных материалов (см. Алкидные смолы), смолы, модифицированные фурфури-ловым спиртом (а также немодифицир. М.-ф. с.),-как связующие в литейном произ-ве при получении отливок из чугуна, стали и алюминия. В качестве пигментов в произ-ве бумаги, добавок для эластомеров, адсорбентов разл. масел и орг. продуктов применяют п о л и м е т и л е н м о ч е в и н у-аморфный или кристаллич. нерастворимый порошкообразный продукт белого цвета, образующийся в сильнокислой среде при взаимод. мочевины и формальдегида (молярное соотношение 1:1).
Первые продукты конденсации мочевины с формальдегидом получены в 1896, производство смол налажено лишь в 1920-21.
Терефталевая кислота+этиленгликоль=полиэфир полиэтилентерефталат
ПОЛИЭТИЛЕНТЕРЕФТАЛАТ, кристаллизующийся полиэфир ф-лы I, мол. м. (20-50)·103. Степень кристалличности и зависит от способа получения полиэтилентерефталата. Аморфный полиэтилентерефталат-твердый прозрачный с серовато-желтоватым оттенком, кристаллический-твердый непрозрачный бесцветный. Ниже приведены нек-рые св-ва полиэтилентерефталата:

Электрич. св-ва полиэтилентерефталата при т-рах до 180°С и в присут. влаги изменяются незначительно. Мех. св-ва определяются способом переработки (напр., волокна, пленки).
Полиэтилентерефталат не раств. в воде и многих орг. р-рителях, раств. лишь при 40-150 °С в фенолах и их алкил- и хлорзамещенных, анилине, бензиловом спирте, хлороформе, пиридине, ди-хлоруксусной и хлорсульфоновой к-тах, циклогексаноне и. др. Для оценки мол. массы методом вискозиметрии используют р-ры полиэтилентерефталата в техн. смеси крезолов, о-хлорфеноле, смеси фенолтетрахлорэтана (1:1) и др. Обладает низкой гигроскопичностью (водопоглощение обычно 0,4-0,5%), к-рая зависит от фазового состояния полимера и относит. влажности воздуха. В УФ области практически непрозрачен. Подвергается фотохим. деструкции, но в меньшей степени, чем поли-e-капроамид.
Характеризуется высокой термостойкостью расплава (~290°С); деструкция на воздухе начинается при т-ре на 50 °С ниже, чем в инертной среде. Эксплуатац. св-ва сохраняются в диапазоне от — 60 до 170°С.
Для повышения термо-, свето-, огнестойкости, для изменения цвета, фрикционных и др. св-в в полиэтилентерефталат вводят разл. добавки (см. ниже), используют также методы хим. модифицирования (разл. дикарбоновыми к-тами и гликолями, к-рые вводят при синтезе полиэтилентерефталата в реакц. смесь).
Получают полиэтилентерефталат поликонденсацией терефталевой к-ты или ее диметилового эфира с этиленгликолем по периодич. или непрерывной схеме в две стадии. По техн.-экономич. показателям преимущества имеет непрерывный процесс получения полиэтилентерефталата из к-ты и этиленгликоля. Этерификацию к-ты этиленгликолем (молярное соотношение компонентов от 1:1,2 до 1:1,5) проводят при 240-270°С и давлении 0,1-0,2 МПа. Полученную смесь бис-(2-гидроксиэтил)тере-фталата с его олигомерами подвергают поликонденсации в нескольких последовательно расположенных аппаратах, снабженных мешалками, при постепенном повышении т-ры от 270 до 300 °С и снижении разряжения от 6600 до 66 Па. После завершения процесса расплав полиэтилентерефталата выдавливается из аппарата, охлаждается и гранулируется или направляется на формование волокна. Матирующие агенты (TiO2), красители, инертные наполнители (каолин, тальк), антипирены, термо- и светостабилизаторы и др. добавки вводят во время синтеза или в полученный расплав полиэтилентерефталата. Перерабатывают полиэтилентерефталат чаще всего экструзией.
Применяют полиэтилентерефталат гл. обр. для произ-ва волокон и нитей (см. Полиэфирные волокна), пленок, бутылей, упаковочного материала, контейнеров и др.
Мировое произ-во полиэтилентерефталата в 1989 составило ок. 9,3 млн. т, причем 90% всего полиэтилентерефталата расходуется на произ-во волокон.
Впервые волокнообразующий полиэтилентерефталат был синтезирован в Великобритании в 1941.
№6. Получение полиорганосилоксанов.
Силикон (полиорганосилоксан) — кислородосодержащее высокомолекулярное кремнийорганическое соединение, обладающее рядом уникальных качеств в комбинациях, отсутствующих у любых других известных веществ: способности увеличивать или уменьшать адгезию, придавать гидрофобность, работать и сохранять свойства при экстремальных и быстроменяющихся температурах или повышенной влажности, диэлектрические свойства, биоинертность, химическая инертность, эластичность, долговечность, экологичность. Это обуславливает их высокую востребованность в разных областях строительства.
1) Получение полиорганосилоксанов с линейными цепями молекул (органосилоксановых эластомеров):
Полиорганосилоксаны с линейными цепями молекул
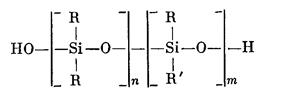
где 1) R и R' — метил; 2) R — метил, a R' — винил; 3) R — метил, a R' — фенил и др., получают реакциями гидролитической поликонденсации или полисоконденсации дифункциональных органохлор-силанов.
Дифункциональные органохлорсиланы при гидролизе проявляют большую склонность к циклизации. Так, например, диметилдихлорсилан гидролизуется водой (в отсутствие растворителя) по конденсационно-полимеризационному механизму с образованием смеси диметилсилоксанов линейного и циклического строения:
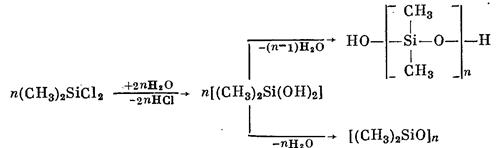
Образование циклических соединений возрастает с величиной органических.радикалов, связанных с кремнием. Например, метил-фенилдихлорсилан и дифенилдихлорсилан образуют при гидролизе преимущественно циклические продукты. В процессе образования циклов при гидролизе органодихлорсиланов важную роль играют условия реакции, в частности рН среды. С увеличением рН, т. е. с уменьшением кислотности среды, можно уменьшить процесс образования циклов, но полностью избежать его невозможно.
Поэтому при получении полидиорганосилоксанов с линейными цепями молекул важнейшей реакцией является полимеризация циклов, образующих-ся при гидролитической поликонденсации диорганодихлорсиланов. Для раскрытия молекул органоциклосилоксанов и получения линейных полиоргано-силоксанов используют реакцию каталитической полимеризации. В качестве инициаторов полимеризации применяются инициаторы катионного типа H2S04, Н3В03, Н3Р04, НООС—СООН, BF3 и др. и анионного типа, например NaOH, КОН, R3SiONa, и др.
При катионной полимеризации, например, с серной кислотой процесс заключается в следующем: на начальной стадии инициирования при взаимодействии органоциклосилоксана с серной кислотой протон кислоты атакует атом кислорода силоксанового цикла. В результате перераспределения электронной плотности связь Si—О разрывается с раскрытием цикла и образованием активного центра на конце цепи:
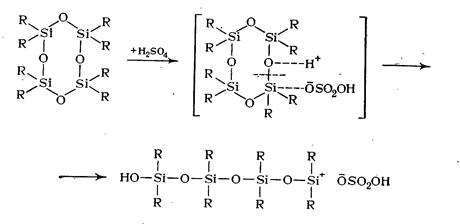
Образовавшийся активный центр ведет дальнейший процесс полимеризации (рост цепи), сопровождающийся размыканием следующих циклов:
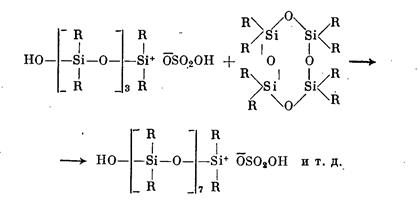
Превращение циклов в линейную полимерную цепь продолжается до достижения равновесия в системе. Обрыв цепи связан с переносом заряда при взаимодействии макрокатиона с молекулами серной кислоты или с захватом макрокатионом присутствующих в системе анионов:
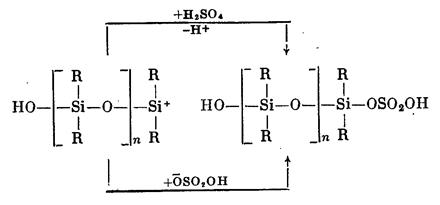
В случае полимеризации органоциклосилоксанов в присутствии анионных инициаторов, например сс-окси-ю-тетраметиламмоний-оксидиметил силок сана, анион взаимодействует с атомом кремния. При этом происходит координационное связывание нуклеофильного реагента с циклом, ослабление кремний-кислородной связи и раскрытие цикла:
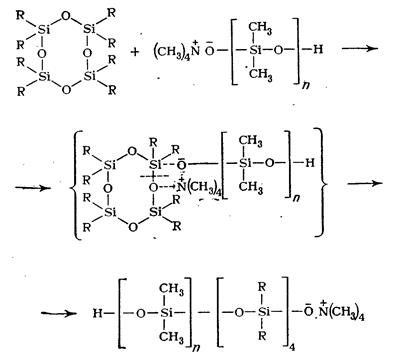
Образующийся активный центр взаимодействует далее со следующей циклической молекулой, раскрывая ее:
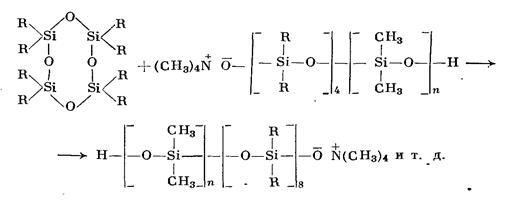
Превращение циклов в линейную полимерную цепь происходит и в этом случае в результате взаимодействия активного центра со следующими циклическими молекулами и тоже продолжается до достижения равновесия.
Обрыв цепи в обоих случаях каталитической полимеризации происходит при потере активности, т. е. при потере концевыми группами способности присоединять циклические молекулы. Это может быть следствием отщепления концевых групп — путем омыления сульфатных групп водой (в случае катионной полимеризации) или путем термической деструкции тетраметиламмониевых групп (в случае анионной полимеризации).
В настоящее время выпускается несколько марок органосилоксановых эластомеров: полидиметилсилоксан (СКТ), полидиметилметилвинилсилокса-ны (СКТВ и СКТВ-1, различающиеся содержанием метилвинил-силоксизвеньев), полидиметилдиэтилсил-оксан (СКТЭ), полидиметилме-тилфенилсилоксан (СКТФ), низкомолекулярные полидиметилсилоксаны (СКТН и СКТН-1, различающиеся молекулярным весом), полиметилфе-нилсилоксан (СКТМФ) и др.
2) Получение полиорганосилоксанов с разветвленными и циклическими цепями
Полиорганосилоксаны разветвленного I и лестничного II строения получают гидролитической поликонденсацией трифункциональных кремнийорганических соединений (лестничные полимеры) или смеси ди- и трифункциональных соединений (разветвленные полимеры).
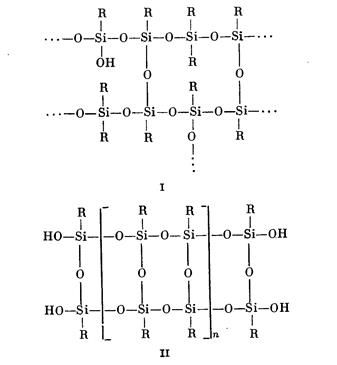
Решающее влияние на свойства полиорганосилоксанов разветвленного и лестничного строения оказывают два фактора: функциональность исходных мономеров, определяемая соотношением числа органических групп или радикалов в них к атому кремния и степень использования функциональных групп в процессе синтеза полимеров. Когда соотношение R: Si снижается с 2 до 1, полимеры постепенно делаются менее текучими, плавкими и растворимыми — в зависимости от эффективности сшивания. При R:Si = 1, т. е. когда в качестве исходного сырья применяются лишь трифункцио- нальные мономеры (метилтрихлорсилан, фенилтрихлорсилан или смесь метил- и фенилтрихлорсиланов), образуются жесткие полимеры, а это значит, что их растворы в органических веществах (лаки) образуют при отверждении трехмерную жесткую структуру. При полном использовании функциональных групп получаются в основном неплавкие и нерастворимые сшитые продукты; однако при том же самом соотношении R: Si специальные методы обработки исходных органотрихлорсиланов могут приводить к лестничным структурам молекул с образованием гибких высокоплавких или неплавких, но растворимых продуктов.
Введение в основную цепь полимера дифункциональных звеньев (диметилсилокси-, диэтилсилокси- или метилфенилсилоксизвеньев) приводит к образованию эластичных полимеров циклолинейной структуры:
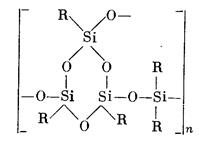
Таким образом, с увеличением соотношения R: Si и уменьшением числа сшивок можно получить полимеры от стеклообразных до каучукоподобных. Большинство полимеров циклолинейного и разветвленного строения получается при соотношении R: Si в пределах 1,0—1,6.
Большое влияние на свойства полиорганосилоксанов оказывает и природа органических групп R, обрамляющих атомы кремния. Увеличение длины алкильных радикалов делает полимер более мягким, повышает его растворимость в органических растворителях и гидрофобизирующую способность, но уменьшает стойкость к термоокислительной деструкции и нагреванию; фенильные радикалы повышают термостойкость полимера. Широкое распространение получили полиорганосилоксаны, содержащие фенильные и метальные группы в обрамлении главной цепи молекулы.
Наряду с такими положительными свойствами полиорганосилоксанов, как высокие тепло- и морозостойкость, хорошая гидрофобность и повышенные диэлектрические характеристики, они обладают недостаточно высокими физико-механическими показателями. Для улучшения этих качеств их часто модифицируют различными органическими полимерами (полиэфирными, эпоксидными и др.).
В настоящее время промышленность выпускает большой ассортимент полиорганосилоксанов разветвленного, циклолинейного и лестничного строения, различающихся по типу органических групп или радикалов, стоящих у атома кремния. Процесс производства таких полиорганосилоксанов основан на реакциях гидролиза или согидролиза органохлорсиланов и последующей поликонденсации продуктов согидролиза.
Основные классы полиорганосилоксанов разветвленного, циклолинейного и лестничного строения такие:
полиметилсилоксаны;
полифенилсилоксаны;
полидиметилфенилсилоксаны и полиметилфенилсилоксаны;
полидиэтилфенилсилоксаны;
5) полиалкилсилоксаны с алкильными радикалами С4 и более у атома Si.
кремнийорганический полиорганосилоксан молекула согидролиз
№7. Реакции полиприсоединения на примере синтеза полиуретанов.
ПОЛИПРИСОЕДИНЕНИЕ (аддиционная поликонденсация. миграционная полимеризация), поликонденсация. не сопровождающаяся выделением низкомол. веществ. Типичные примеры полиприсоединёние-образование полиуретанов из диизоцианатов и гликолей (ур-ние 1) и полимочевин из диизоцианатов и диаминов (2), отверждение диэпоксидов диаминами (3) или диолами:
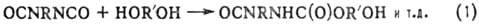
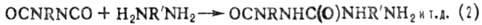
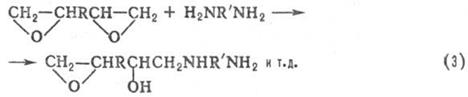
Полиуретаны получают взаимодействием соединений, содержащих изоцианатные группы с би- и полифункциональными гидроксилсодержащими производными. В качестве изоцианатов используются толуилендиизоцианаты (2,4- и 2,6-изомеры или их смесь в соотношении 65:35), 4,4'-дифенилметандиизоцианат, 1,5-нафтилен-, гекса-метилендиизоцианаты, полиизоцианаты, трифенилметан-триизоцианат, биуретизоцианат, изоциануратизоцианаты, димер 2,4-толуилендиизоцианата, блокированные изоцианаты. Строение исходного изоцианата определяет скорость уретанообразования, прочностные показатели, световую и радиационную стойкость, а также жёсткость полиуретанов. Гидроксилсодержащими компонентами являются:
олигогликоли — продукты гомо- и сополимеризации Тетрагидрофурана, пропилен- и этиленоксидов, дивинила, изопрена; сложные полиэфиры с концевыми группами ОН — линейные продукты поликонденсации адипиновой, фталевой и других дикарбоновых кислот с этилен-, пропилен-, бутилен- или другими низкомолекулярным гликолями; разветвленные продукты поликонденсации перечисленных кислот и гликолей с добавкой триолов (глицерина, триметилол-пропана), продукты полимеризации ε-капролактона. Гидроксилсодержащий компонент определяет, в основном, комплекс физико-механических свойств полиуретанов. Для удлинения и структурирования цепей применяются гидроксилсодержащие вещества (например, вода, гликоли, моноаллиловый эфир глицерина, касторовое масло)и диамины (-4,4'-метилен-бис-(о-хлоранилин), фенилен-диамины). Эти агенты определяют молекулярную массу линейных полиуретанов, густоту вулканизационной сетки и строение поперечных химических связей, возможность образования доменных структур, то есть комплекс свойств полиуретанов и их назначение (пенопласты, волокна, эластомеры и т. д.). В качестве катализаторов для процесса уретанообразования используют третичные амины, хелатные соединения железа, меди, бериллия, ванадия, нафтенаты свинца и олова, октаноат и лауринат олова. При процессе циклотримеризации катализаторами являются неорганические основания и комплексы третичных аминов с эпоксидами.
Образование полиуретанового полимера путем реакции между диизоцианатом и полиолом:
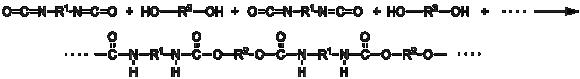
№9 Способы проведения полимеризации и поликонденсации:
Способы проведения поликонденсации
Выбор способа проведения поликонденсация определяется физико-химическими свойствами исходных веществ и образующихся полимеров, технологическими требованиями, задачами, которые ставятся при осуществлении процесса, и т.д. По температуре способы проведения поликонденсация делят на высокотемпературные и низкотемпературные (см. табл.), по агрегатному состоянию реакционной системы или фазовому состоянию - на поликонденсацию в массе (расплаве), твердой фазе, растворе, эмульсии (суспензии), двухфазной системе (межфазная поликонденсация). Поликонденсация в расплаве и твердой фазе происходит при высоких температурах, поликонденсация в эмульсии и межфазная поликонденсация - при низких температурах, поликонденсация в растворе - при высоких и низких температурах. Низкотемпературная поликонденсация является преимущественно неравновесной, высокотемпературная - преимущественно равновесной.
СРАВНЕНИЕ МЕТОДОВ НИЗКОТЕМПЕРАТУРНОЙ И ВЫСОКОТЕМПЕРАТУРНОЙ ПОЛИКОНДЕНСАЦИИ
| Характеристика | Низкотемпературная ПОЛИКОНДЕНСАЦИЯ | Высокотемпературная ПОЛИКОНДЕНСАЦИЯ |
| Мономеры | ||
| Чистота | От средней до высокой | Высокая |
| Стехиометрич. соотношение | Часто допустимы определенные отклонения | Обязательно |
| Термич. стойкость | Необязательна | Необходима |
| Химическая строение | Разнообразное; ограничено требованием реакционное способности | Ограничено термостойкостью; пониж. требования к реакционное способности |
| Условия поликонденсации | ||
| Продолжительность | Неск. мин | 1-24 ч |
| Температура, 0C | 0-50 | Не ниже 200 |
| Давление | Атмосферное | Высокое и низкое |
| Оборудование | Простое и открытое | Иногда весьма сложное (часто автоклавное) |
| Продукты реакции | ||
| Полимер | ||
| выход | От низкого до высокого | Высокий |
| молекулярная масса | (50-100)•103 и выше | (20-30)• 103, иногда выше |
| химический строение | Разнообразное | Ограничено термостойкостью |
| Побочный продукт | Соль | Вода или летучие органическое соединения |
Методы проведения реакции полимеризации весьма разнообразны.
Блочная полимеризация. При блочной полимеризации, иначе называемой полимеризацией в массе, полимер содержит минимальное количество примесей. Однако по мере протекания реакции повышается вязкость системы, ухудшаются условия теплопередачи (на стадии роста реакция является экзотермической), что приводит к получению продуктов с пониженным молекулярным весом и широким молекулярно-весовым распределением (высокой полидисперсностью). Вообще говоря, любой полимерный продукт практически является полидисперсным, однако слишком большая полидисперсность часто нежелательна, особенно при синтезе пленкообразователей; фракции с низкой степенью полимеризации ухудшают физико-механические показатели последних, а слишком высокомолекулярные фракции плохо растворимы.
Лаковая полимеризация. Полимеризация в растворе, или лаковая полимеризация, осуществляется в жидкой среде, которая является растворителем как для мономера, так и для образующегося полимера. При этом облегчается теплообмен, однако растворитель может участвовать в реакции передачи цепи, вызывая снижение среднего молекулярного веса. Лаковые полимеризаты, как правило, не используются в качестве связующих по той причине, что из них трудно удалить не вступивший в реакцию мономер (практически реакции полимеризации никогда не протекают со 100%-ным выходом полимера). Присутствие же остаточного мономера в полимеризационном лаковом связующем понижает его стабильность. Кроме того, мономеры часто бывают токсичными (например, мономеры акрилового ряда). Поэтому полимер высаждают из полимеризата в виде твердого осадка, который отделяют, промывают (иногда переосаждают), высушивают и уже затем растворяют в подходящем растворителе (часто совсем не в том, в котором производилась лаковая полимеризация).
Суспензионная полимеризация. Известны два метода полимеризации диспергированных в воде мономеров: суспензионная (капельная) и эмульсионная (латексная). Основное преимущество проведения полимеризации в эмульсии (как и в суспензии) заключается в возможности использования дисперсионной среды для регулирования отвода тепла, выделяющегося при полимеризации. Однако механизмы протекания полимеризационных процессов в эмульсии и суспензии существенно различны, поэтому образуются продукты с разными свойствами. Неодинаков и характер получаемых водных дисперсий полимеров.
| <== предыдущая | | | следующая ==> |
| Еңбекті қорғау және өртке қарсы іс –шаралар | | | Особенности создания Российской империи |
Date: 2015-07-27; view: 8023; Нарушение авторских прав