Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Л. А. Животовский
|
|
генетическая история человечества
ВВЕДЕНИЕ
При изучении генетической истории человечества первоочередная задача – это выявление глобальной демографической картины в истории человечества и датировка основных событий: распространения человека по планете, разделение на различные ветви (географические группы, или расы), и рост численности разных групп популяций.
Последние годы изучение эволюционной истории человечества проводили на основе анализа специфических участков генома человека: митохондриальной ДНК (мтДНК) и Y-хромосомных маркеров. Основное преимущество их в относительной простоте анализа, но принципиальная их важность – в том, что они позволяют исследовать раздельно материнскую и отцовскую линии, поскольку мтДНК передается потомкам только от матери, а Y-хромосома наследуется только по отцовской линии. Этот анализ позволил многое выявить в эволюционном прошлом человечества, однако ряд важных деталей выявленной с их помощью картины эволюции человечества находятся в противоречии друг с другом. Например, ретроспективный анализ изменчивости ДНК в мировых популяциях показал, что все ныне существующие типы мтДНК сводятся к т.н. «митохондриальной Еве», а типы Y-хромосомы – к «Y-Адаму», причем разного возраста. Несомненно, большая часть этих кажущихся противоречий заключается в том, что и мтДНК и Y-хромосома представляют лишь малые части генома человека, причем части, по-разному отражающие миграционные события и возможно подверженные отбору.
Поэтому сейчас встала важная проблема изучения, наряду с мтДНК и Y-хромосомой, большого числа аутосомных маркеров, представляющих все хромосомы человека. Кроме того, анализ аутосомных ДНК-маркеров позволил бы свести вместе комбинативное действие материнских и отцовских линий, поскольку такие маркеры наследуется и по материнской, и по отцовской линиям. Такой подход позволил бы получить гораздо более полную картину эволюции популяций человека. В данной работе описываются результаты такого подхода, впервые примененного для глобального анализа древней демографической истории популяций человека путем генетического исследования ныне существующих народов.
Наиболее перспективными генетическими маркерами для подобного эволюционного анализа являются микросателлитные локусы. Микросателлиты представляют собой фрагменты ДНК с повторяющимися «мотивами» – идентичными короткими последовательностями из нескольких нуклеотидов (например, тетрануклеотидный мотив GATA), откуда их другое название – STR-локусы (от англ. Short Tandem Repeats). Индивиды могут отличаться друг от друга по числу повторов в каждом STR-локусе. STR-локусы многочисленны: они распределены по всему геному, и сейчас картированы сотни локусов на каждой хромосоме человека (Weber and Broman 2001). Они отличаются высокой скоростью мутирования (Dib et al. 1996), порядка 0.1% и выше на локус на поколение, что позволяет эффективно исследовать эволюционные процессы на коротких промежутках времени.
Что чрезвычайно важно, данные по изменчивости STR-локусов хорошо описываются развитым математическим аппаратом современной популяционной генетики. Это привело к созданию математико-статистических методов анализа и моделированию эволюционного процесса. Среди математического инструментария – RST -статистики популяционной дифференциации (Slatkin 1995), генетическая дистанция (δμ)2 (Goldstein et al. 1995), TD -статистика для оценки времени дивергенции (разделения) популяций (Zhivotovsky 2001), высшие моменты вероятностных распределений по числу повторов (Zhivotovsky and Feldman 1995). Эти методы позволяют провести ретроспективную оценку роста численности древних популяций человека по данным о микросателлитах в современных популяциях (Kimmel et al. 1998, Reich and Goldstein 1998, Gonser et al. 2000, Jin et al. 2000, King et al. 2000, Zhivotovsky et al. 2000) и оценку интенсивности миграций между соседними популяциями (Slatkin 1995, Michalakis and Excoffier 1996, Rousset 1996, Feldman et al. 1999).
МАТЕРИАЛЫ И МЕТОДЫ
Для исследования глобальной картины генетической эволюции человечества были исследованы 1056 человек из 52 популяций, биологические образцы которых хранятся в виде клеточных культур (the HGDP-CEPH Human Genome Diversity Cell Line Panel, Cann et al. 2002). Эти образцы были генотипированы по 404 STR-локусам в The Mammalian Genotyping Service (см. http://research.marshfieldclinic.org/genetics/sets/ combo. html, Marshfield Panel #10), охватывающим все хромосомы, включая половые хромосомы; из них мы выбрали 377 аутосомных STR: 45 с ди-, 58 с три- и 274 с тетра-нуклеотидными повторами. Эти данные сейчас доступны по адресу http://research.marshfieldclinic.org/genetics/Freq/FreqInfo.htm. (Данные по X- и Y-хромосоме еще не проанализированы нами полностью и потому в этом исследовании не рассматриваются).
Выборки индивидов охватывали популяции (этнические группы) всех населенных континентов (в этой статье названия этнических групп даны на английском – так, как они были обозначены в указанных выше Интернет-сайтах, # означает номер). А именно, Южная Африка: охотники-собиратели племени пигмеев Биака (Biaka, Центральная африканская республика, #47) и Мбути (Mbuti, Конго, #48) и племени Сан (San, Намибия, #50), а также оседлых народов – «фермеров», занимающихся сельским хозяйством, которые мы будем здесь иногда обозначать собирательным термином «Банту» – Bantu (Кения, #49), Yoruba (Нигерия, #51) и Mandenka (Сенегал, #52); Северная Африка: Mozabite (Алжир, #44); Ближний Восток: друзы (Druze, Израиль, Кармел, #41), палестинцы (Palestinian, Центральный Израиль, #42), бедуины (Bedouin, Израиль, Негев, #43); Центральная/Южная Азия: уйгуры (Uygur, Северо-Западный Китай, #20) и племена из Пакистана – Balochi (#24), Brahui (#25), Burusho (#26), Hazara (#27), Kalash (#28), Makrani (#29), Pathan (#30), Sindhi (#31); Европа: баски (Basques, Франция, #33), французы (Франция, #34), Bergamo (#35), Sardinian (#36), Tuscan (#37) – все из Италии, Orcadian (о-ва Оркни, #38), русские (Север Европейской части России, #39), адыги (Adygei, Северный Кавказ, #40); Восточная Азия: Cambodian (Камбоджа, #6), а также выборки Dai (#7), Daur (#8), Han из Северного Китая (#9) и из США (#10), Hezhen (#11), Lahu (#12), Miao (#13), монголы (Mongola, #14), Naxi (#15), Oroqen (#16), She (#17), Tu (#18), Tujia (#19), Xibo (#21), Yi (#22) – все из Китая, японцы (Япония, #23), якуты (Сибирь, #32); Океания: меланезийцы (Melanesian, Боганвил, #45), папуасы (Papuan, Новая Гвинея, #46); Америка: южно-американские племена бассейна Амазонки Karitiana (Бразилия, #1) и Surui (Бразилия, #2) и колумбийцы (Colombian, Колумбия, #3), Центрально-американские народности майя (Maya, Мексика, #4) и Pima (Мексика, #5). Объемы выборок были небольшие, средний размер был около 20; детальное описание дано в Rosenberg et al. (2002).
Было обнаружено, что изученные STR-локусы значительно отличаются друг от друга как по числу аллелей (рис. 1), так и по дисперсии числа повторов (рис. 2) – вне зависимости от типа локуса (с ди-, три- или тетрануклеотидными повторами). Это может быть связано как с различной скоростью мутирования в разных локусах (Zhivotovsky et al. 2001), так и с генетической «ошибкой» – стохастичностью траекторий эволюционного процесса (Weir 1996).
Мы определили 52 х 52-матрицу парных значений FST (точнее, θ, Weir 1996), используя компьютерную программу GDA (Lewis and Zaykin 2001), на основе которой определили главные координаты (PC) путем многомерного шкалирования с использованием компьютерной программы SPSS 8.0.0. Для оценки времени дивергенции популяции применили TD –статистику (Zhivotovsky 2001); необходимую для TD –статистики частоту мутаций по микросателлитным локусам определяли по Zhivotovsky et al. (2000) с модификацией Zhivotovsky et al. (2003). Для оценки времени начала роста популяций в численности применяли индекс дисбаланса –ln 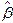 (Kimmel et al. 1998; King et al. 2000) и индекс экспансии Sk (Zhivotovsky et al. 2000). Показатель Sk позволяет также оценить численность пра-популяции перед тем, как она стала расти в численности в предположении, что перед этим пра-популяция находилась в генетическом равновесии (уравнения 7, 8 из Zhivotovsky et al. 2000). Оценки Sk интерпретируются как минимальные границы времени экспансии (начала роста численности), реальное время экспансии более раннее, если численность растет медленно и/или темпы мутирования сильно варьируют от локуса к локусу. Для моделирования генетической динамики популяций мы использовали программу “Mathematica” (Wolfram 1996). Мы также исследовали распределение приватных (популяционно-специфических) аллелей, т.е. аллелей, которые встречаются только в этой выборке индивидов и больше ни в какой другой, которые могут дать полезную информацию о популяционно-генетической струтуре (см. Barton and Slatkin 1986). Были введены две статистики: S3 = S1/N и S4=100 S1 S2 / L (в процентах). Здесь S1 – это суммарное число в данной выборке различных приватных аллелей по всем локусам, S2 – это средняя частота приватных аллелей в выборке, а N – объем выборки.
(Kimmel et al. 1998; King et al. 2000) и индекс экспансии Sk (Zhivotovsky et al. 2000). Показатель Sk позволяет также оценить численность пра-популяции перед тем, как она стала расти в численности в предположении, что перед этим пра-популяция находилась в генетическом равновесии (уравнения 7, 8 из Zhivotovsky et al. 2000). Оценки Sk интерпретируются как минимальные границы времени экспансии (начала роста численности), реальное время экспансии более раннее, если численность растет медленно и/или темпы мутирования сильно варьируют от локуса к локусу. Для моделирования генетической динамики популяций мы использовали программу “Mathematica” (Wolfram 1996). Мы также исследовали распределение приватных (популяционно-специфических) аллелей, т.е. аллелей, которые встречаются только в этой выборке индивидов и больше ни в какой другой, которые могут дать полезную информацию о популяционно-генетической струтуре (см. Barton and Slatkin 1986). Были введены две статистики: S3 = S1/N и S4=100 S1 S2 / L (в процентах). Здесь S1 – это суммарное число в данной выборке различных приватных аллелей по всем локусам, S2 – это средняя частота приватных аллелей в выборке, а N – объем выборки.
РЕЗУЛЬТАТЫ
Выделение популяционных кластеров. Метод многомерного шкалирования позволил выделить группы генетически связанных друг с другом популяций (Рис. 3, 4). Наиболее важным здесь оказалось то, что популяции из одного и того географического региона кластеризуются вместе. Можно выделить следующие пять групп: Южная Африка, Западная Евразия, Восточная Азия, Океания и Америка. Эти группы представляют собой основные расы человека, выделяемые антропологами по морфологическим признакам.
Различные координаты по-разному дифференцируют популяции. В проекции главных координат PC1 и PC2 выделяются три больших группы: Африка/Западная Евразия, Восточная Азия/Океания и Америка (Рис. 3). Южная Африка ясно выделяется координатой PC4, а Океания - координатой PC5 (Рис. 4), хотя их отделение (правда, гораздо менее четкое) проявляется по первым двум координатам (Рис. 3). Эти результаты согласуются с нашим анализом этих же данных, проведенных другим методом (Rosenberg et al. 2002).
Положение отдельных популяций в пределах каждого кластера соответствует их региональной группировке. Например, популяции «фермеров» Южной Африки ближе друг к другу, чем к племенам охотников-собирателей San и Mbuti (#50 и #48), Рис. 3; последние лежат на границе южно-африканского кластера, обособляясь от другого племени пигмеев, Biaka (#47), оказавшегося в под-кластере популяций фермеров, что имет свое объяснение (см. Обсуждение).
Западная Евразия, которая включает Ближний Восток, Европу, Центральную и Южную Азию и Северную Африку, четко отделяется от других больших групп и имеет выраженную внутреннюю структуру. Так, Северная Африка (представленная в нашей базе данных единственной выборкой Mozabite) лежит на краю этого генетического кластера. Популяции Ближнего Востока группируются вместе. Популяции из Европы также образуют собственный суб-кластер. Однако Basques (#33), Sardidnian (#36) и Orcadian (#38) больше тяготеют к ближневосточному суб-кластеру. Kalash, популяция из Пакистана, и возможно имеющая корни на Ближнем Востоке или даже в Европе, лежит в европейском суб-кластере (рис. 3). Другие популяции Центральной и Южной Азии образуют свой генетический суб-кластер на рис. 3. Uygur, Hazara и Burusho (## 20, 27, 26) занимают положение, промежуточное между Евразией и Восточной Азией, особенно Uygur и Hazara, что, по-видимому, отражает их генеалогическое родство с монголами.
Этнические группы Восточной Азии формируют отдельный генетический кластер (рис. 3), в котором также прослеживается определенная внутренняя структура. Популяции Lahu, She, Naxi, и Miao южного Китая тяготеют к нижнему краю восточно-азиатского кластера, в то время как большинство популяций северного Китая, принадлежащие алтайской языковой семье (Daur, Hezhen, Mongola, Oroqen, Tu, и Xibo), расположены в верхней части кластера. Океания также отчетливо отделяется от остальных географических групп (рис. 4). И наконец, популяции Америки также явно отделяются от других континентальных групп и, кроме того, показывают гораздо бóльшую внутрирегиональную генетическую дифференциацию, чем популяции других регионов (рис. 3). Одна из амазонских популяций, а именно Surui (#2), вообще обособлена от других американских популяций, вероятно из-за значительного генетического дрейфа, вызванного чрезвычайно малой численностью этого племени. Другое племя бассейна реки Амазонка, Karitiana (#1), также генетически значительно отличается от остальных популяций. Центрально-американская популяция майя (#4), напротив, показывает бóльшую, чем другие американские популяции, близость к африканским и европейским популяциям, что может отражать влияние миграций на американский континент в пост-Колумбову эпоху.
Оценки времени дивергенции основных эволюционных линий и времени начала роста численности популяций. Для этой цели мы воспользовались TD -статистикой, которая позволяет оценить время, когда пара популяций отделилась от общей пра-популяции, и статистку ∆TD для оценки времени, прошедшего между соседними точками ветвления на популяционном древе (Zhivotovsky 2001). Полученное популяционное древо, вместе с временами ответвления основных эволюционных ветвей, представлено на рис. 5. Наиболее древняя ветвь представлена африканскими охотниками-собирателями; от них отделяется ветвь, ведущая к современным популяциям Банту и вне-южноафриканским популяциям. В свою очередь последняя последовательно ветвится на Западную Евразию, Океанию, Восточную Азию и Америку с более дробным делением в пределах каждой из них.
Статистика TD имеет то преимущество перед другими мерами генетического расстояния, что ее статистическая оценка не зависит от того, как меняется численность популяций во времени, и устойчива к возможным слабым миграционным потокам. Это очень важное ее преимущество, поскольку популяции всегда меняются в численности, а человечество в целом росло и растет. В то же время, для ее применения необходимо знать величину V0 – генетическую изменчивость (дисперсию числа повторов) в предковой популяции (Zhivotovsky 2001). Однако она неизвестна, что приводит к неопределенности в оценке TD. Тем не менее, эта неопределенность может быть заключена в нижнюю и верхнюю границы через задание соответствующих границ для V0. Действительно, приравняв V0 нулю, мы получаем величину TD =141.7 ± 5.7 тыс. лет, которая является верхней границей для времени первой дивергенции – отделения от «древней» ветви, ныне представленной популяциями охотников-собирателей, ветви, ведущей к «фермерам» Южной Африки. С другой стороны, взяв величину V0 большей по величине, чем она была в древней африканской пра-популяции, мы получили бы нижнюю границу для времени первой дивергенции. Как выбрать такую величину V0? Для этого можно попытаться найти среди ныне существующих популяций такие, которые могли бы стать генетической моделью древней африканской пра-популяции. Можно допустить, что изолированные популяции бассейна Амазонки (Karitiana и Surui) представляют собой такую модель, поскольку они, возможно, следуют тому образу жизни и поддерживают ту численность, которая была характерна для самых первых древних орд человека. (Некоторые дополнительные аргументы в пользу выбора этих популяций в качестве модели рассмотрены в Обсуждении). Взяв в качестве V0 величину дисперсии в популяциях Karitiana и Surui, мы оценили нижнюю границу возраста корня глобального древа, т.е. время первого деления на рис. 5, как 71.2 ± 4.4 тыс.лет. Полученные здесь верхняя и нижняя границы дают почти в два раза более узкий предел, чем полученный ранее – 50 и 200 тыс.лет (Harpending et al. 1998).
Оценки времени отделения других ветвей популяционного древа рис 5 на основе показателя ∆TD не требуют знания величины дисперсии числа повторов в точке ответвления, однако при применении этого показателя предполагается, что генетическая изменчивость в соседних точках ветвления одинакова. Если же популяции растут в численности в промежутке времени между точкам ветвления (точнее, если растет дисперсия числа повторов), то ∆TD дает нижнюю границу времени, прошедшего между двумя соседними ветвлениями; если бы дисперсия падала, тогда бы ∆TD давала верхнюю границу времени (Zhivotovsky 2001). Поскольку человечество в целом росло, то ∆TD дает нижнюю границу времени между соседними ветвлениями популяционного древа. В Обсуждении мы даем дополнительные аргументы в пользу того, что внутри-популяционная дисперсия числа повторов растет вдоль ветвей древа. Следовательно, все оценки времени между ветвлениями на рис. 5 должны рассматриваться как их нижние границы – фактические их величины не меньше, чем указанные на рис. 5.
Далее, мы вычислили индекс дисбаланса –ln (Kimmel et al. 1998) и индекс экспансии Sk (Zhivotovsky et al. 2000) для определения того, росли ли популяции человека или нет: для растущих популяций эти индексы положительны – Табл. 2. Сами эти статистики дают сопоставляемые результаты (коэффициент корреляции между ними – 0.86). Однако имеются и некоторые различия: индекс дисбаланса указывает на рост численности популяций охотников-собирателей Южной Африки (правда, этот индекс не дает критерия значимости отличия от нуля, так что, возможно, это значение индекса незначимо), в то время как индекс экспансии не отличается статистически значимо от нуля. Значимо положительные величины Sk для «фермеров» Южной Африки и популяций Западной Евразии и Восточной Азии (Табл. 2) указывают на то, что эти популяции устойчиво росли в численности. Популяции Океании и Америки не несут генетических следов устойчивого роста численности. Табл. 2 показывает, что популяции фермеров Южной Африки стали увеличиваться в численности раньше, чем популяции Западной Евразии и Восточной Азии и что их эффективная численность была до момента роста невелика – менее двух тысяч, что соответствует физической численности в шесть тысяч, согласно правилу «утроения» (Cavalli-Sforza et al. 1994). Интерпретация всех этих фактов дана в Обсуждении.
ОБСУЖДЕНИЕ
Генетические расстояния и методы кластеризации. Реконструкция популяционной истории должна опираться на генетическую информацию, отражающую изменчивость всего генома (Barbujani and Bertorelle 2001), что обеспечивается вовлечением в анализ большого числа локусов (Zhivotovsky and Feldman 1995, Goldstein et al. 1996, Jorde et al. 1997). Если в эволюционных исследованиях изучать только небольшую часть существующего разнообразия, то степень разрешения методов анализа может быть невелика и положение ветвей на популяционном дереве может оказаться неточным. Наше исследование опирается на информацию о маркерах, представляющих все хромосомы, и табл. 3 говорит о том, что увеличение числа изучаемых маркеров уменьшает стандартные ошибки оценок времени дивергенции популяций. Анализ, проведенный здесь (рис. 3, 4) и ранее (Rosenberg et al. 2002), показывает, что несколько сот STR-локусов стопроцентно различают континентальные группы популяций друг от друга.
Другие исследования также показывают, что популяции одного региона располагаются близко на популяционном дереве (напр., Cavalli-Sforza et al. 1988, Nei and Roychoudhury 1993, Bowcock et al. 1994, Deka et al. 1995, Takezaki and Nei 1996, Jorde et al. 1997, Pérez-Lezaun et al. 1997, Jin et al. 2000, Watkins et al. 2001), топология которого зависит от того, какие меры генетического расстояния и методы кластеризации использованы. Мы сравнили четыре типа деревьев, построенных методами ближайшего соседа и UPGMA на основе матрицы 52x52 величин FST и RST и обнаружили, что, как правило, на каждом из них популяции одного региона группируются вместе. Однако генные потоки, популяционная динамики, отклонения от генетического равновесия и другие нарушения предположений, на которых основана интерпретация генетических расстояний, могут привести к смещению оценок времени дивергенции и неверной топологии популяционного дерева. Важно подчеркнуть, что наши оценки времени дивергенции, основанные на TD, слабо зависят от популяционной динамики и, в отличие от оценок, основанных на других мерах генетического расстояния, не предполагают генетического равновесия популяций.
Возможные сценарии эволюции популяций человека. Укажем наиболее важные особенности генетической изменчивости популяций из различных регионов мира, данные в табл. 1-2 и рис. 5. Во-первых, популяции охотников-собирателей Южной Африки имеют наибольшую генетическую изменчивость (дисперсию числа повторов) – бόльшую, чем другие популяции, в том числе и популяции «фермеров» Южной Африки. И это несмотря на то, что их численность (в среднем, около 50 тысяч, см. Cavalli-Sforza et al. 1994) несравненно ниже, чем популяции «фермеров» (миллионы людей); теоретически, в равновесной популяции генетическая изменчивость прямо пропорциональна численности. Кроме того, в ходе эволюции популяции охотников-собирателей Южной Африки не отличались устойчивым ростом: индекс экспансии, тестирующий устойчивый популяционный рост, незначимо отличается от нуля для популяций охотников-собирателей (табл. 3). Во-вторых, гораздо более многочисленные популяции Азии показывают меньшее генетическое разнообразие, чем африканские популяции (как охотников-собирателей, так и фермеров). В-третьих, индекс экспансии не показывает никакого роста численности популяций Океании и Америки. Более того, хотя нативные популяции Америки имели экологические возможности для роста численности до нашествия белых людей, генетическая изменчивость в них наименьшая среди изученных популяций. В четвертых, наблюдаемое уменьшение генетической изменчивости в направлении «Африка-Азия-Океания-Америка» полностью соответствует дивергенции соответствующих популяционных групп на популяционном древе (рис. 5). В-пятых, как следует из теории (Slatkin 1995, Zhivotovsky and Feldman 1995), для того чтобы обеспечить наблюдаемую максимальную величину генетической изменчивости (напр. 3.45 для STR-локусов с тетрануклеотидными повторами у охотников-собирателей Южной Африки), достаточно иметь эффективную численность, не превышающую 2700, т.е. неизмеримо меньшую, чем численность даже малых аборигенных популяций Южной Африки.
Сколь мала была по численности древняя пра-популяция Южной Африки? Предполагая, что время первого ветвления популяционного древа (возраст корня) – это среднее значение нижней и верхней границ, данных на рис. 5, и что эта предковая популяция была в генетическом равновесии, ее эффективная численность оценивается как 700. Эта оценка, соответствующая данным Pritchard et al. (1999), Zhivotovsky et al (2000) и Tang et al. (2002), меньше по величине, чем оценки, полученные в других исследованиях (напр., 10000 в работе Harpending et al. 1998). Однако полученная нами цифра не отвергает возможности того, что в Африке в то время могли существовать многие популяции Homo sapiens sapiens, но при этом утверждает, что, скорее всего, они были генетически изолированы друг от друга и что современные народы произошли от одной или от малого числа тех древних популяций. Недавние генетические исследования южно-африканских племен, в языке которых имеются щелкающие звуки, показали, что вероятно именно они являются потомками той древней предковой популяции, от которой произошло все современное человечество (Knight et al. 2003).
Что происходило с внутрипопуляционной генетической изменчивостью в ходе эволюции? Согласно данным выше расчетам, современные популяции охотников-собирателей имеют бόльшую эффективную численность, чем древняя африканская предковая популяция, а это значит, что генетическая изменчивость могла неуклонно увеличиваться по мере роста численности этих популяций за прошедшее длительное время. (С другой стороны, эти популяции росли достаточно медленно и, вероятно, флуктуировали в численности, как это характерно для ряда аборигенных народов. Поскольку индекс экспансии более чувствителен к колебаниям численности, чем генетическая изменчивость (дисперсия числа повторов) (Zhivotovsky et al. 2000), то он не достигает значимого уровня в африканских популяциях охотников-собирателей). Аналогичным образом, рост численности в других региональных группах популяций также влек за собой увеличение внутрипопуляционной генетической изменчивости во времени в каждой из этих групп.
Однако темпы роста внутрипопуляционной изменчивости, вероятно, разнились в разных региональных группах. Действительно, в ныне существующих популяциях наблюдается четкий географический тренд в уменьшении генетической изменчивости (табл. 2): от максимальной величины в африканских популяциях и затем все меньшая изменчивость в ряду Азия-Океания-Америка. Этот тренд можно объяснить тем, что каждая новая ветвь популяционного дерева образовывалась небольшой по численности группой основателей. Эта группа несла достаточно изменчивости, чтобы не вызвать эффекта «генетического горлышка бутылки», но в то же время была малочисленной и уменьшала скорость роста этой изменчивости во времени. Поэтому достигшие большей численности более древние популяции Африки наращивали генетическую изменчивость быстрее, чем более молодые популяции Азии, а эти, в свою очередь, – быстрее, чем еще более молодые популяции Америки. Рост численности в популяциях Океании, и, следовательно, рост их генетической изменчивости, мог сдерживаться еще и условиями существования в этом регионе.
Заметим, что эффективная численность древней африканской предковой популяции (~700) могла обеспечить дисперсию числа повторов, равную 2 х 700 х 0.00064 = 0.90 (по тетрануклеотидным локусам), что гораздо меньше наблюдаемой дисперсии в минимальных по изменчивости амазонских популяциях (Karitiana и Surui) – 1.8. Следовательно, дисперсия числа повторов в древней африканской предковой популяции десятки тысяч лет назад была меньше, чем в ныне существующих амазонских популяциях. Этот расчет является дополнительным аргументом в пользу выбора амазонских популяций как модели генетической изменчивости древней пра-популяции: их генетическая дисперсия может быть взята в качестве верхней границы V0 при датировке первого ветвления популяционного дерева в глубоком прошлом человечества (рис. 5).
Низкая численность древней предковой популяции может также объяснить, почему современные популяции человека не столь генетически разнообразны, как другие родственные виды. Например, разнообразие мтДНК всех людей гораздо меньше разнообразия мтДНК в небольшой группе шимпанзе (Gagneux et al. 1999). Следовательно, эволюционная линия, ведшая к древней предковой популяции, должна была быть небольшой численности (см. Gagneux et al. 1999, Yang 2002), а эволюционного времени, прошедшего со времени экспансии популяций человека, оказалось недостаточно для формирования большого разнообразия. Наши данные (табл. 2) о времени формирования основных популяционных групп (африканские популяции фермеров, Западная Евразия и Восточная Азия) находятся в соответствии с недавними исследованиями (Jorde et al. 1997, Di Rienzo et al. 1998, Excoffier and Schneider 1999, Gonser et al. 2000, Jin et al. 2000, Shen et al. 2000), хотя в ряде работ не отмечалось генетических следов роста численности за пределами Африки (Reich and Goldstein 1998, Jin et al. 2000). В некоторых работах предполагается наличие эффекта «горлышка бутылки» (Kimmel et al. 1998, Watkins et al. 2001, Gabriel et al. 2002), хотя этот эффект и подразделение на субпопуляции может одинаково сказываться на статистических тестах экспансии (Frisse et al. 2001, Reich et al. 2001, Pluzhnikov et al. 2002).
Древность популяций охотников-собирателей Южной Африки. П опуляционное дерево рис.5 подтверждает теорию африканского происхождения человека современного анатомического типа, расселившегося по миру через цепочку миграций на Ближний Восток, Европу, Азию, Океанию и Америку. Это качественно соответствует результатам, основанным на данных о мтДНК, Y-хромосоме и аутосомных микросателлитов с динуклеотидными повторами (напр., Bowcock et al. 1994, Ingman et al. 2000, Underhill et al. 2000), хотя не исключен вклад архаичного человека в современный пул генов. Наш анализ указывает на то, что охотники-собиратели Южной Африки являются потомками тех, кто был в корне популяционного дерева, т.е. они потомки наиболее древних популяций (рис. 5). Сказанное подтверждается данными о распределении «приватных» (private) аллелей, т.е. аллелей, которые встречаются только в данной популяции и больше ни в какой другой. Оказалось, что чаще всего приватные аллели есть среди охотников-собирателей, а после них – в популяциях «фермеров» Южной Африки (табл. 4). Это соответствует наблюдению, что популяционно-специфичные гаплотипы, выделяемые комбинациями внутригенных SNP, наиболее часты у афро-американцев, по сравнению с белым и испано-язычным населением Америки (Stephens et al. 2001). Еще одним аргументом в пользу древности популяций охотников-собирателей Южной Африки является максимальное разнообразие аллелей в этих популяциях, в т.ч. внутри-популяционная дисперсия числа повторов: 3.45 ± 0.119 (табл. 2). Хотя эта величина статистически незначимо превышает таковую для популяций «фермеров» (3.31 ± 0.108), она гораздо выше, чем для не-африканских народов: 2.90±0.035, 2.61±0.036, 2.27±0.100, и 2.11±0.070 для Западной Евразии, Восточной Азии, Океании и Америки, соответственно.
Какие из ныне живущих народов Африки являются самыми древними, – это проблема, требующая детального исследования. Анализ RFLP- и SNP-маркеров митохондриальной ДНК (Chen et al. 2000) и полных последовательностей мтДНК (рис. 2 в Ingman et al. 2000) показывает, что San и Mbuti входят в один генетический кластер, а Biaka – в другой. Согласно нашим данным (рис. 5), племена San и Mbuti представляют, вероятно, древнейшие ветви человечества среди изученных здесь популяций. Не исключено, что именно San – наиболее древняя ветвь, хотя различие между временами отделения San, а затем Mbuti по нашим данным статистически незначимо. Эволюционная ветвь, ведущая к племени Biaka, вероятно, отделилась значительно позднее и она гораздо ближе к следующей по времени ветви «фермеров» (Банту) Южной Африки (рис. 4 и 5), хотя на интерпретации наших данных мог сказаться значительный генный поток между популяциями Банту и Biaka (Cavalli-Sforza 1986). Другим генетическим свидетельством древности эволюционной ветви, ведущей к San, являются статистики приватных аллелей (табл. 4), которые достигают максимальных значений именно в популяции San. Далее, наиболее древние линии Y-хромосомы встречаются с высокой частотой именно в популяции San (Underhill et al. 2000, Cruciani et al. 2002, Semino et al. 2002). И, наконец, язык San, включающий щелкающие звуки, вероятно, самый древний среди ныне существующих языков (Knight et al. 2003).
Популяции охотников-собирателей Южной Африки не несут генетических следов устойчивого роста их численности в древние времена: индекс экспансии Sk незначителен и незначимо отличается от нуля (табл. 2), что находится в соответствии с заключением Excoffier и Schneider (1999). В отличие от них, популяции Банту имеют ясно выраженные следы роста численности. Анализ моделей динамики роста дисперсии V и индекса Sk привел к заключению, что наблюдаемые значения этих статистик могут быть объяснены тем, что популяции фермеров-банту стали расти в численности, по крайней мере, 35 тыс. лет назад, когда их эффективная численность была лишь около 2000 (табл. 2), что соответствовало физической численности племен около 6000. Вероятно, данная величина это нижняя граница времени экспансии, поскольку вариации в скорости мутирования могли уменьшить эту цифру и поскольку метод оценки предполагает «скачок» численности: при постепенном росте динамика исследованных показателей замедляется.
Охота и простое собирательство не допускало этого, в результате чего популяции охотников-собирателей «застыли» в своей численности (см., Cavalli-Sforza et al. 1994, p. 106). В тоже время, рост численности африканских популяций «фермеров» мог быть обусловлен техническими достижениями того времени – например, улучшением каменных орудий труда, датируемым примерно 50 тыс. лет (Cavalli-Sforza et al. 1994, p. 64), что хорошо соответствует приведенной выше нижней границе в 35 тыс. лет для экспоненциального роста в эволюционной ветви Банту, а также прежними оценками времени начала роста численности популяций Южной Африки: 70 тыс. лет (Excoffier and Schneider 1999) и 60 тыс. лет (Zhivotovsky et al. 2000).
Заселение других континентов. Каждая из больших популяционных групп (южно-африканские «фермеры», Западная Евразия, Восточная Азия) является мета-популяцией, т.е. состоящей из популяций с определенным уровнем генетического обмена и с общим происхождением. Это подтверждается статистикой приватных аллелей S4, величина которой для объединенных данных по всей системе гораздо выше, чем ее усредненное значение для отдельных популяций (табл. 4). (Сравним эти данные с величинами S4 для африканских охотников-собирателей: 0.89 для объединенных данных San, Biaka и Mbuti против среднего значения 0.85 для S4 в каждой из этих популяций в отдельности; такое соотношение означает их относительную изоляцию друг от друга).
Согласно нашим оценкам, ветвь, ведущая к современным популяциям Океании, сформировалась в период образования популяций Центральной и Южной Азии (рис. 5); археологические находки подтверждают, что человек появился в Новой Гвинее примерно 40–60 тыс. лет назад. Популяции Океании содержат наибольшее число приватных аллелей среди всех не-африканских популяций (табл. 4). Одно из возможных объяснений этого – несколько волн древних миграций из Африки, одна из которых направилась в Океанию (см. Jin et al. 1999) – с последующей длительной изоляцией популяций Океании от континента и, напротив, обменом мигрантами между континентальными популяциями
Наиболее спорным в современной теории эволюции человека – это время заселения Америки: предполагается несколько волн миграций на континент через Берингию от 12 до 40 тыс. лет тому назад. Поскольку археологические данные свидетельствуют об увеличении численности стоянок около 12-ти тыс. лет назад (Nettle 1999) и поскольку индекс экспансии для американских популяций не показывает роста численности (табл. 2), можно предположить, что общий рост численности населения Америки был связан с увеличением числа небольших по объему популяций с незначительным генетическим обменом между ними. Вероятно, некоторые популяции Америки прошли через «бутылочное горлышко»; по крайней мере, это произошло с племенем Surui, для которого индекс экспансии наименьший среди всех изученных нами популяций: Sk = –0.221±0.138. Хотя значение индекса экспансии статистически незначимо отличается от нуля, имеется еще ряд свидетельств тому: Surui наиболее отклоняющаяся из всех популяций в координатах, полученных многомерным шкалированием генетических расстояний (рис. 4), а внутрипопуляционная дисперсия в Surui наименьшая среди всех изученных нами популяций – 1.68±0.139. Кроме того, был обнаружен аллель (под номером 275) тетрануклеотидного локуса D9S1120, который представлен во всех изученных популяциях Америки и отсутствует в не-американских популяциях. Этот аллель практически фиксирован в Surui (частота 0.97), вероятно вследствие генетического дрейфа, и достигает величин 0.2-0.3 в остальных популяциях Америки: 0.30 в Maya, 0.22 в Pima, 0.19 в Colombia, 0.25 в Karitiana. Столь широкое распространение этого аллеля в пределах Америки означает, что он возник среди основателей аборигенного населения Америки.
Указанный аллель 275 локуса D9S1120 можно рассматривать как генетический маркер для популяций Америки. Это наименьший по числу повторов аллель из обнаруженных по этому локусу. Помимо этого аллеля обнаружен еще один в этом же локусе, также встречающийся только в популяциях Америки – на один повтор больше (279). Следующие по размеру аллели (283 и 287) обнаружены в разных популяциях мира, не только в Америке, но частота их очень низкая.
В других популяциях и региональных группах по изученным локусам не были найдены столь четкие специфичные маркеры, как ка маркер 275 для популяций Америки, хотя некоторые приватные аллели были достаточно частые. Например, в Южной Африке имеются два приватных для этого региона аллеля, частота которых превышает 10%, в то время как в популяциях охотников-собирателей имеются свои собственные приватные аллели, частота одного из которых около 16%. В популяции San обнаружено большое число приватных аллелей, два из которых превышают 30%. В популяциях Океании обнаружены два частых приватных аллеля с частотой превышающей 10%, а в каждой из ее популяций (New Guinea и Melanesia) имеются свои собственные относительно частые приватные аллели. В отличие от этого, в Западной Евразии и Восточной Азии приватные аллели редки и не превышают 3%. В популяциях южно-африканских фермеров-банту ни один из приватных аллелей не достигает частоты выше 5%, за исключением одного – около 8%. Ни в одной из указанных выше популяций или региональных групп не найдено маркера, который бы отделил их – однако, эти группы выделяются комбинациями сотен не-приватных аллелей (Rosenberg et al. 2002, и рис. 3-4 данного исследования).
Наши данные демонстрируют очень сложную популяционную структуру и сложную популяционную историю трех наиболее крупных групп популяций – южно-африканских «фермеров», Западной Евразии и Восточной Азии. Это хорошо видно по распределению приватных аллелей: если различные пары популяций или групп популяций объединить, то в них появляются дополнительные, специфические для этих пар приватные аллели. Например, в популяциях южно-африканских охотников-собирателей и фермеров-банту обнаружено, соответственно, 160 и 100 приватных аллелей. Однако если их «слить», то общее число приватных аллелей возрастет до 321; таким образом, имеются 61 дополнительных приватных аллелей, характерных только для выборок из Южной Африки. То же самое обнаружено для не-африканских популяционных групп: Центральной/Южной Азии и Восточной Азии, и Центральной/Южной Азии и Европы (32 и 24 дополнительных аллеля, соответственно). Вероятно это обусловлено миграционными потоками, что находится в соответствии с данными Karafet et al. (1999) и Wells et al. (2001) о том, что центрально-азиатские популяции являются источником миграций в Восточную Азию и в Европу. Аналогичный анализ распределения приватных аллелей показал, что популяции Ближний Востока и Северной Африки аналогичным образом связаны с популяциями Европы и Азии. Анализ приватных аллелей позволяет также выявить сильную связь между популяциями Южной Африки и популяциями Ближнего Востока/Северной Африки, Центральной/Южной Азии и Восточной Азии, что соответствует гипотезе обратных миграций в Африку по данным анализа Y-хромосомных линий (Cruciani et al. 2002), хотя не исключено, что дифференциальная потеря аллелей вследствие генетического дрейфа также внесла свой вклад в наблюдаемую картину распределения приватных аллелей.
В заключение статьи укажем, что изучение распределения в популяциях географически и этнически ограниченных приватных аллелей и аллелей, распространенных широко, т.е. обнаруживаемых в различных этнических группах, в т.ч. географически удаленных друг от друга, позволяет выявить различные стороны популяционной истории: первые представляют генные потоки между популяциями, в то время как широко распространенные аллели позволяют обрисовать глобальную демографическую картину давнего прошлого в истории человечества. Наше исследование обнаруживает четкую генетическую дифференциацию основных региональных групп популяций – Южной Африки, Западной Евразии, Восточной Азии, Океании и Америки – (рис. 3, 4), представляющих большие расы, и выявляет эволюционные взаимосвязи между ними (рис. 5).
ЛИТЕРАТУРА
Andrews P (1986) Fossil evidence on human origins and dispersal. In: Cold Spring Harb. Symp. Quant. Biol. 51: 419-428
Barbujani G, Bertorelle G (2001) Genetics and the population history of Europe. Proc Natl Acad Sci USA 98: 22-25
Barton, NH, Slatkin M (1986) A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 56: 409-415
Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL. 1994. High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368: 455-457
Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, Chen Z (2002) A human genome diversity cell line panel. Science: 296: 261-262
Cann RL, Stoneking M, Wilson AC (1987) Mitochondrial DNA and human evolution. Nature 325: 31-36
Carvalho-Silva D.R., Santos, F.R., Rocha, J. & Pena, S.D.J. (2001). The phylogeography of Brazilian Y-chromosome lineages. Am J Hum Genet 68: 281-286
Cavalli-Sforza, L.L. (1986) African Pygmies. Academic Press, Orlando, Florida.
Cavalli-Sforza LL,Menozzi P, Piazza A (1994) The History and Geography of Human Genes. Princeton Univ. Press. Princeton, N.J.
Cavalli-Sforza LL, Piazza A, Menozzi P, Mountain J (1988) Reconstruction of human evolution: bringing together genetic, archaeological, and linguistic data. Proc Natl Acad Sci USA 85: 6002-6006
Chakraborty R, Kimmel M, Stivers DN, Davison LJ, Deka R (1997) Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc Natl Acad Sci USA 94: 1041-1046
Chen FC, Li WH (2001) Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet 68: 444-456
Chen YS, Olckers A, Schurr TG, Kogelnik AM, Huoponen K, Wallace DC (2000) mtDNA variation in the South African Kung and Khwe: and their genetic relationships to other African populations. Am J Hum Genet 66: 1362-1383
Cruciani F, Santolamazza P, Shen PD, Macaulay V, Moral P, Olckers A, Modiano D, Holmes S, Destro-Bisol G, Coia V (2002) A back migration from Asia to sub-Saharan Africa is supported by high-resolution analysis of human Y-chromosome haplotypes. Am J Hum Genet. 70: 1197-1214
Deka R, Jin L, Shriver MD, Yu LM, DeCroo S, Hundrieser J, Bunker CH, Ferrell RE, Chakraborty R (1995) Population genetics of dinucleotide (dC–dA)n·(dG–dT)n polymorphisms in world populations. Am J Hum Genet 56: 461-474
Dib C, Faure S, Fizames C et al. (14 co-authors) (1996). A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380: 152-154
Di Rienzo A, Donnelly P, Toomajian CH, Sisk B, Hill A, Petzl-Erler ML, Haines GK, Barch DH (1998) Heterogeneity of microsatellite mutations within and between loci, and implications for human demographic histories. Genetics 148: 1269-1281
Excoffier L, Schneider S (1999) Why hunter-gatherer populations do not show signs of Pleistocene demographic expansions. Proc Natl Acad Sci USA 96: 10597-10602
Feldman MW, Kumm J, Pritchard JK (1999) Mutation and migration in models of microsatellite evolution. In: Goldstein DG, Schlotterer C (eds.), Microsatellites: Evolution and Applications. Oxford Univ. Press 98-115
Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A (2001) Gene conversion and different population histories may explain the contrast metween polymorphism and linkage disequilibrium levels. Am J Hum Genet 69: 831-843
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J et al. (18 co-authors) (2002) The structure of haplotype blocks in the human genome. Science 296: 2225-2229
Gagneux P, Wills C, Gerloff U, Tautz D, Morin PA, Boesch C, Fruth B, Hohmann G, Ryder OA, Woodruff DS (1999) Mitochondrial sequences show diverse evolutionary histories of African hominoids. Proc Natl Acad Sci USA 96: 5077-5082
Goldstein DB, Zhivotovsky LA, Nayar K, Linares AR, Cavalli-Sforza LL, Feldman MW (1996) Statistical properties of the variation at linked microsatellite loci: implications for the history of human Y chromosomes. Mol Biol Evol 13: 1213-1218
Goldstein DB, Linares AR, Cavalli-Sforza LL, Feldman MW (1995) Genetic absolute dating based on microsatellites and the origin of modern humans. Proc Natl Acad Sci USA 92: 6723-6727
Gonser R, Donnelly B, Nicholson G, Di Rienzo A (2000) Microsatellite mutations and inferences about human demography. Genetics 154: 1793-1807
Harpending HC, Batzer MA, Gurven M, Jorde LB, Rogers AR, Sherry ST (1998) Genetic traces of ancient demography. Proc Natl Acad Sci USA 95: 1961-1967
Hey J (1998) Population genetics and human origins - haplotypes are key! Trends in Genet 14: 303-304
Horai S, Maruyama K, Hayasaka K, Matsubayashi S, Hattori Y, Fusharoen G, Harihara S, Park KS, Omoto K, Pan IH (1996) mtDNA polymorphism in East Asian populations, with special reference to the peopling of Japan. Am J Hum Genet 59: 579-590
Hurles ME, Irven C, Nicholson J, Taylor PG, Santos FR, Loughlin J, Jobling MA, Sykes BC (1998) European Y-chromosomal lineages in Polynesians: A contrast to the population structure revealed by mtDNA. Am J Hum Genet 63: 1793-1806
Ingman M, Kaessmann H, Paabo S, Gyllensten U (2000) Mitochondrial genome variation and the origin of modern humans. Nature 408: 708-713
Jin L, Underhill PA, Doctor V, Davis RW, Shen PD, Cavalli-Sforza LL, Oefner PJ (1999) Distribution of haplotypes from a chromosome 21 region distinguishes multiple prehistoric human migrations. Proc Natl Acad Sci USA 96: 3796-3800
Jin L., Bakett ML, Cavalli-Sforza LL, Zhivotovsky LA, Feldman MW, Rosenberg NA. 2000. Microsatellite evolution in modern humans: a comparison of two data sets from the same populations. Annals Hum Genet 64: 117-134
Jorde LB, Rogers AR, Bamshad M, Watkins WS, Krakowiak P, Sung S, Kere J, Harpending H. 1997. Microsatellite diversity and the demographic history of modern humans. Proc Natl Acad Sci USA 94: 3100-3103
Karafet T, Xu L, Du R, Wang W, Feng S, Wells RS, Redd AJ, Zegura SL, Hammer MF (2001) Paternal population history of East Asia: Sources, patterns, and microevolutionary processes. Am J Hum Genet 69: 615-628
Kayser M, Krawczak M, Excoffier L, Dieltjes P, Corach D, Pascali V, Gehrig C, Bernini LF, Jespersen J, Bakker E, Roewer, de Knijff P (2001) An extensive analysis of Y-chromosomal microsatellite haplotypes in globally dispersed human populations. Am J Hum Genet 68: 990-1018
Kimmel M, Chakraborty R, King JP, Bamshad M, Watkins WS, Jorde LB (1998) Signatures of population expansion in microsatellite repeat data. Genetics 148: 1921-1930
King JP, Kimmel M, Chakraborty R (2000) A power analysis of microsatellite-based statistics for inferring past population growth. Mol Biol Evol 17: 1859-1868
Knight, A., P.A. Underhill, H.M. Mortensen, L.A. Zhivotovsky, A.A. Lin, B.M. Henn, D. Louis, M. Ruhlen, and J.L. Mountain. 2003. African Y chromosome and mtDNA divergence provides insights into the history of click languages. Current Biology 13: 464-473
Lewis PO, Zaykin D (2001) Genetic Data Analysis: Computer program for the analysis of allelic data. Version 1.0 (d16c). Free program distributed by the authors over the internet from http://lewis.eeb.uconn.edu/lewishome/software.html
Michalakis Y, Excoffier L (1996) A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142: 1061-1064.
Moran PAP (1975) Wandering distributions and the electrophoretic profile. Theor. Popul. Biol. 8: 318-330
Nei M, Roychoudhury AK (1993) Evolutionary relationships of human populations on a global scale. Mol Biol Evol 10: 927-943
Nei M, Takezaki N (1996) The root of the phylogenetic tree of human populations. Mol Biol Evol 13: 170-177
Nettle D (1999) Linguistic diversity of the Americas can be reconciled with a recent colonization. Proc Natl Acad Sci USA 96: 3325-3329
Pérez-Lezaun A, Calafell F, Mateu E, Comas D, Ruiz-Pacheco, Betranpetit J (1997) Microsatellite variation and the differentiation of modern humans. Hum Genet 99: 1-7
Pluzhnikov A, Di Rienzo A, Hudson RR (2002) Inferences about human demography based on multilocus analyses of noncoding sequences. Genetics 161: 1209-1218
Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman MW (1999) Population growth of human Y chromosomes: A study of Y chromosome microsatellites. Mol Biol Evol 16: 1791-1798
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945-959
Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC et al. (11 co-authors) (2001) Linkage disequilibrium in the human genome. Nature 411: 199-204.
Reich DE, Goldstein DB. 1998. Genetic evidence for a Paleolithic human population expansion in Africa. Proc Natl Acad Sci USA 95: 8119-8123
Relethford JH (2001) Genetics and the Search for Modern Human Origins. Wiley-Liss, New York
Reynolds J, Weir BS, Cockerham CC (1983) Estimation of the co-ancestry coefficient: basis for a short-term genetic distance. Genetics 105: 767-779
RibeirodosSantos AKC, Guerreiro JF, Santos SEB, Zago MA (2000) The split of the Arara population: Comparison of genetic drift and founder effect. Hum. Heredity 51: 79-84
Rickards O, MartinezLabarga C, Lum JK, DeStefano GF, Cann RL (1999) mtDNA history of the Cayapa Amerinds of Ecuador: Detection of additional founding lineages for the native American populations. Am J Hum Genet 65: 519-530
Rogers AR and Jorde LB (1996) Ascertainment bias in estimates of average heterozygosity. Am J Hum Genet 58: 1033-1041
Rosenberg NA, Pritchard JK, JL Weber, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW (2002) Genetic structure of human populations. Science (in press)
Rousset, F (1996) Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics 142: 1357-1362
Ruiz-Linares A, Minch E, Meyer D, Cavalli-Sforza LL (1995) Analysis of classical and DNA markers for reconstructing human population history. In The Origins and Past of Homo Sapiens Sapiens as Viewed From DNA. Hanihara K, Brenner S, eds. World Scientific Publishers, Singapore
Semino O, Passarino G, Oefner PJ, et al. (17 co-authors) (2000) The genetic legacy of paleolithic Homo sapiens sapiens in extant Europeans: A Y chromosome perspective. Science 290: 1155-1159
Semino O, Santachiara-Benerecetti AS, Falaschi F, Cavalli-Sforza LL, Underhill PA (2002) Ethiopians and Khoisan share the deepest clades of the human Y-chromosome phylogeny. Am J Hum Genet 70: 265-268
Shen P, Wang F, Underhill PA, Franco C, Yang W-H, Roxas A, Sung R, Lin AA, Hyman RW, Vollrath D, Davis RW, Oefner PJ (2000) Population genetic implications from sequence variation in four Y chromosome genes. Proc Natl Acad Sci USA 97: 7354-7359
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457-462
Stephens JC, Schneider JA, Tanguay DA, et al. (29 co-authors) (2001) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293: 489-493
Stringer CB, Andrews P (1988) Genetic and fossil evidence for the origin of modern humans. Science 239: 1263-1268
Takezaki N, Nei M (1996) Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144: 389-399
Tang H., Thomson R, Cavalli-Sforza LL, Shen P, Oefner P, Feldman MW. Sex differences in demographic histories of humans. (Submitted)
Urbanek M, Goldman D, Long JC (1996) The apportionment of dinucleotide repeat diversity in native Americans and Europeans: A new approach to measuring gene identity reveals asymmetric patterns of divergence. Mol Biol Evol 13: 943-953
Underhill PA, Shen P, Lin AA, et al. (21 co-authors) (2000) Y chromosome sequence variation and the history of human populations. Nature Genet 26: 358-361
Watkins WS, Ricker CE, Bamshad MJ, Caroll ML, Nguen SV, Batzer MA, Harpending HC, Rogers AR, Jorde LB (2001) Patterns of ancestral human diversity: An analysis of Alu-insertion and restriction-size polymorphisms. Am J Hum Genet 68: 738-752
Weber JL, Broman KW (2001) Genotyping for human whole-genome scans: past, present, and future. Advances in Genet 42: 77-96
Weir BS (1996) Genetic Data Analysis II. Methods for Discrete Population Genetic Data. Sinauer Ass., Sunderland, Mass.
Wells RS, Yuldasheva N, Ruzibakiev R, Underhill PA, Evseeva I, Blue-Smith J, Jin L, et al. (2001) The Eurasian heartland: A continental perspective on Y-chromosome diversity. Proc Natl Acad Sci USA 98: 10244-10249
Wilson JF, Weiss DA, Richards M, Thomas MG, Bradman N, Goldstein DB (2001) Genetic evidence for different male and female roles during cultural transitions in the British Isles. Proc Natl Acad Sci USA 98: 5078-5083
Wolfram S (1996). Mathematica. A System for Doing Mathematics by Computer. (3rd ed.) Wolfram Media/Cambridge Univ. Press, NY
Yang Z (2002) Likelihood and Bayes estimation of ancestral population sizes in hominoids using data from multiple loci. Genetics 162: 1811-1823
Zhivotovsky LA (2001) Estimating divergence time with the use of microsatellite genetic distances: impacts of population growth and gene flow. Mol Biol Evol 18: 700-709
Zhivotovsky LA, Bennett L, Bowcock AM, Feldman MW (2000) Human population expansion and microsatellite variation. Mol Biol Evol 7: 757-767
Zhivotovsky LA, Feldman MW (1995) Microsatellite variability and genetic distances. Proc Natl Acad Sci USA 17: 11549-11552
Zhivotovsky LA, Goldstein DB, Feldman MW (2001) Genetic sampling error of distance (δμ)2 and variation in mutation rate among microsatellite loci. Mol Biol Evol 18: 2141-2145
Zhivotovsky LA, Rosenberg N, Feldman MW (2003) Features of evolution and expansion of modern humans inferred from genome-wide microsatellite markers. Amer. J. Hum. Genet. (in press)
Date: 2015-07-10; view: 363; Нарушение авторских прав