Полезное:
Как сделать разговор полезным и приятным
Как сделать объемную звезду своими руками
Как сделать то, что делать не хочется?
Как сделать погремушку
Как сделать так чтобы женщины сами знакомились с вами
Как сделать идею коммерческой
Как сделать хорошую растяжку ног?
Как сделать наш разум здоровым?
Как сделать, чтобы люди обманывали меньше
Вопрос 4. Как сделать так, чтобы вас уважали и ценили?
Как сделать лучше себе и другим людям
Как сделать свидание интересным?

Категории:
АрхитектураАстрономияБиологияГеографияГеологияИнформатикаИскусствоИсторияКулинарияКультураМаркетингМатематикаМедицинаМенеджментОхрана трудаПравоПроизводствоПсихологияРелигияСоциологияСпортТехникаФизикаФилософияХимияЭкологияЭкономикаЭлектроника
Fig. 1
Structures of silybin and taxifolin.
The effect of silybin on cholesterol metabolism was further studied in hypercholesterolemic subjects (9). Although biliary cholesterol concentrations were found to be reduced, the exact compound(s) responsible for this effect was disputed. Krecman et al. (11) studied in rats the hypocholesterolemic effects of both silymarin, a mixture of flavolignans extracted from S. marianum, and pure silybin, a single flavolignan. Interestingly, they found that silybin was not as effective as silymarin, suggesting that other constituent(s) of silymarin, in addition to silybin, may also have hypocholesterolemic effects. Subsequently, the minor constituent of silymarin, taxifolin, has attracted the attention of our laboratory, because taxifolin constitutes the flavonoid moiety in the flavolignan silybin (Fig. 1). Hence, the purpose of our study was to investigate the effects of taxifolin on lipids, apolipoprotein B-100 (apoB), and apolipoprotein A-I (apoA-I) synthesis and secretion, using a well-established human hepatoma cell-line, HepG2, as the model system.
Previous SectionNext Section
MATERIALS AND METHODS
Materials
(±)-Taxifolin (>98% pure; purchased from Sigma, St. Louis, MO) was prepared in ethanol and preserved at –25°C for no longer than 4 weeks. Immediately before use, the stock solution was diluted in culture medium to give a final ethanol concentration of 0.1% (v/v). The concentration used was verified by molar absorptivity (ε = 19,953, 290 nm).
HepG2 cells (HB 8065) were obtained from American Type Culture Collection (ATCC, Rockville, MD). Cell culture media, fetal bovine serum (certified grade), antibiotic-antimycotic mixture, and Trizol reagent were from Life Technologies (Grand Island, NY). Culture dishes and flasks were obtained from Corning Costar (Cambridge, MA). Electrophoresis reagents were from Bio-Rad (Hercules, CA). [35S]protein labeling mix (1,175 Ci/mmol), [1-14C]acetic acid (40–60 mCi/mmol), [1,3-14C]glycerol (>40 mCi/mmol), [1-14C]oleic acid (40–60 mCi/mmol), [ glutaryl -3-14C]hydroxy-3-methylglutaryl coenzyme A (40–60 mCi/mmol), ENHANCE™, and Reflection™ autoradiography films were purchased from NEN Life Science Research Products (Boston, MA). Monospecific goat anti-human apoB antiserum was obtained from Alexon-Trend (Ramsey, MN). Protein A-positive Staphylococcus aureus cells were from Roche Molecular Biochemicals (Indianapolis, IN). Plastic-backed thin-layer chromatography (TLC) plates (Silica Gel 60) were from Alltech Associates (Deerfield, IL). The CytoTox 96 nonradioactive cytotoxicity assay and the pGEM-7Zf vector were from Promega (Madison, WI). Other common laboratory reagents were from Sigma.
Cell culture
Monolayer HepG2 cell cultures were maintained in RPMI 1640 medium with 10% FBS at 37°C with 5% CO2 and 95% air and subcultured in 35-mm-diameter dishes until about 80% confluency. Once confluency was reached, cells were treated with taxifolin in serum-free RPMI (SF-RPMI). Untreated control cells received 0.1% (v/v) ethanol without taxifolin.
Analysis of cellular and secreted lipids
To measure the rates of cholesterol and cholesteryl ester (CE) synthesis and secretion, treated and untreated cells were labeled with [14C]acetate (5 μCi/ml) for 6 h. Triacylglycerol (TAG) and phospholipids, on the other hand, were labeled with [14C]glycerol (2.5 μCi/ml) for 6 h. After labeling, the medium was collected and the cells were washed twice with cold phosphate-buffered saline. Cellular and medium lipids were then extracted with hexane–isopropanol 3:2 (v/v) as described by Goldstein, Basu, and Brown (12). The organic solvent was evaporated, and lipids were resuspended in hexane and spotted on a TLC plate. Neutral and polar lipids were separated using a two-solvent system. Plates were first developed in chloroform–methanol–acetic acid–formic acid–water 70:30:12:4:2 (v/v/v/v/v) and then developed in ether–diethyl ether–glacial acetic acid 90:10:1 (v/v/v). The TLC plates were dried and the lipids were visualized with I2 vapor and zones corresponding to the lipid standard were cut, mixed in scintillation cocktail, and counted on a Packard (Downers Grove, IL) Tri-Carb model 1500 liquid scintillation counter. After lipid extraction, cell proteins were digested in 1 ml of 0.1 M NaOH and measured as described below.
Analysis of HMG-CoA reductase activity
HMGR activity was measured in permeabilized HepG2 cells according to Leonard and Chen (13). Briefly, treated and untreated cells, grown in 24-well culture plates, were permeabilized with digitonin as described by Theriault et al. (14). After permeabilization, cells were rinsed once with cytoskeletal buffer and immediately used for the HMGR assay. The permeabilized cells were first incubated in preincubation buffer [50 mM phosphate buffer (pH 7.4), 10 mM dithiothreitol (DTT), and 1 mM ethylenediaminetetraacetic acid (EDTA)] ± taxifolin for 20 min at 37°C. The enzyme assay was initiated by adding labeling buffer [100 mM phosphate buffer (pH 7.4), 5 mM DTT, 20 mM glucose 6-phosphate, 2.5 mM NADP, glucose-6-phosphate dehydrogenase (3.3 units/ml), and [14C]HMG-CoA (1 μCi/ml, 150 μM)] ± taxifolin for 30 min at 37°C. Finally, the reaction was terminated by the addition of 0.8 M HCl and further incubated for 30 min at 37°C to lactonize mevalonate. The [14C]mevalonolactone formed from [14C]HMG-CoA was isolated by TLC, using a protocol similar to that described above. Briefly, the supernatant was extracted with diethyl ether. After evaporation of the organic solvent, mevalonolactone was resuspended in chloroform – methanol 2:1 (v/v), and an aliquot was spotted on a TLC plate. Mevalonolactone was separated by development in benzene – acetone–acetic acid 400:600:1 (v/v/v) and visualized with I2 vapor. The zone corresponding to the mevalonolactone standard was cut, mixed in a scintillation cocktail, and counted on a scintillation counter. Results (counts per minute) were converted into micromoles per minute per well and expressed relative to the values obtained from untreated control cells. An internal standard, [3H]mevalonate, was added to the assay for estimating recovery.
Analysis of CE formation
Cellular cholesterol esterification was determined according to the method described essentially by Goldstein, Basu, and Brown (12). Briefly, each dish pretreated with or without taxifolin for 22 h received [14C]oleic acid (0.5 μCi/ml, 360 μM) complexed to fatty acid-free bovine serum albumin (BSA) and incubated for a further 2 h in the presence and absence of taxifolin. The molar ratio of oleic acid to BSA was 8:1. The cells were then washed three times with Earle's balanced salt solution (EBSS), the lipids were extracted in situ, and CE was separated by TLC as described above. Cell protein was determined as described below. An internal standard, [3H]cholesterol oleate, was added to the assay to correct for procedural losses.
Metabolic pulse-chase labeling experiments
Treated and untreated HepG2 cells were preincubated in methionine/cysteine-free RPMI for 30 min and pulsed with an [35S]protein labeling medium ([35S]protein labeling mix at 100 μCi/ml in SF-RPMI ± taxifolin) for 10 min. After the short pulse, the cells were washed with EBSS and chased in SF-RPMI 1640 supplemented with 5 mM methionine/cysteine ± taxifolin. At various chase times, duplicate 35-mm dishes were harvested and cells were lysed in solubilization buffer as described previously (14). The lysates were centrifuged for 5 min in a microcentrifuge (7,500 g) and the supernatants were collected for immunoprecipitation. Media collected at each time point were spun as described above to remove any cell debris and mixed with a protease inhibitor cocktail [2 mM phenylmethylsulfonyl fluoride, aprotonin (100 kallikrein-inactivating units/ml), 0.1 mM leupeptin, and N -acetyl-leucyl-leucyl-norleucinal (ALLN, 5 μM final concentration)] prior to immunoprecipitation, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and fluorography as described previously (14).
RNase protection assay for apoB and apoA-I mRNA abundance
ApoB and apoA-I mRNA levels were measured by an RNase protection/solution hybridization assay as described similarly by Azrolen and Breslow (15). This was performed by incubating the labeled probe (3–9 × 104 cpm, 150 pg) with total cellular RNA (20 μg) in 40 μl of hybridization buffer [80% formamide, 40 mM N -2-hydroxyethylpiperazine- N ′-2-ethanesulfonic acid (pH 6.7), 0.4 M NaCl, 1 mM EDTA] overnight at 53°C (for apoB probe) or 63°C (for apoA-I probe). Unhybridized RNA was degraded by the addition of RNase A and RNase T1 in digestion buffer [0.3 mM NaCl, 10 mM Tris-HCl (pH 7.4), 5 mM EDTA] for 1 h at 34°C. Protected RNA was then precipitated with 20% trichloroacetic acid (TCA) and 100 μg of salmon sperm DNA for 15 min on ice. Each sample was filtered through glass fiber filters (GF/C) and washed with 10% TCA. Radioactivity was quantified by counting the filters in a liquid scintillation counter.
Human specific cDNA for apoB was kindly provided by Z. Yao (University of Ottawa, Canada), whereas apoA-I cDNA was purchased from the ATCC. Both amplified fragments were ligated to a pGEM-7Zf vector, which served as the template to synthesize antisense RNA probes. A mouse β-actin construct was used to normalize total RNA amount. Unlabeled cRNA corresponding to the sense DNA strand was prepared for use as an hybridization standard.
Lipoprotein fractionation
Cells were treated and subjected to pulse-chase labeling as described above. The culture medium was collected after the 2-h chase, adjusted to 10% sucrose, and separated by sucrose gradient ultracentrifugation as previously described (14). Centrifugation was carried out at 35,000 rpm at 12°C for 65 h in an SW41 Ti rotor. Gradients were fractionated into 1-ml fractions, and the density and apoB were measured in each fraction. All solutions contained the protease inhibitor cocktail as above.
Other methods
Cell protein content was measured according to Bradford (16) (i.e., Bio-Rad), using BSA as the standard. The activity of lactate dehydrogenase (LDH) released into the medium was measured spectrophotometrically, using the CytoTox 96 nonradioactive cytotoxicity assay according to the manufacturer protocol (Promega).
Statistical analysis
Data were normalized to the amount of cellular protein. Statistical differences were analyzed with a paired t -test with the level of significance set at 0.05.
Previous SectionNext Section
RESULTS
Taxifolin inhibits cholesterol synthesis in a dose- and time-dependent manner
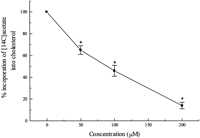
Initial studies were performed to determine an optimal concentration of taxifolin that would inhibit de novo cholesterol synthesis without altering cell viability. As shown in Fig. 2, taxifolin added in various concentrations to the culture medium for 24 h decreased the rate of incorporation of [14C]acetate into cellular cholesterol in a dose-dependent manner. Percent inhibition was 35 ± 4% at 50 μM, 54 ± 5% at 100 μM, and 86 ± 3% at 200 μM. At or below 200 μM, there was no significant release of LDH into the medium, indicating no cytotoxicity effect (data not shown). Furthermore, TCA-precipitable radioactivity from cells incubated with 200 μM taxifolin remained essentially unchanged versus untreated control indicating that the flavonoid did not alter cellular protein synthesis (data not shown). Consequently, this nontoxic pharmacologic dose was chosen in the following experiments. At 200 μM, the decrease in total cellular cholesterol was accompanied by a decrease in both free cholesterol and esterified cholesterol, which reached 85 ± 3 and 80 ± 2% of control, respectively.
Time course studies of HepG2 cells preincubated with 200 μM taxifolin were also performed. In these studies, cells were incubated and labeled with a [14C]acetate labeling medium with and without taxifolin for a total of 6, 18, and 24 h. The inhibitory effect of taxifolin on newly synthesized cholesterol synthesis was found to be time dependent, with an optimal inhibition observed within 24 h (data not shown). In all further experiments, a 24-h treatment was used.
View larger version:
· In this page
· In a new window
· Download as PowerPoint Slide
Date: 2015-09-18; view: 220; Нарушение авторских прав; Помощь в написании работы --> СЮДА... |